

دوره 34، شماره 3 - ( خرداد 1405 )
جلد 34 شماره 3 صفحات 10023-10011 |
برگشت به فهرست نسخه ها


![]()
![]()
![]()
Download citation:
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:



BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
Mojarrad M, Eslahi A, Tanipur H, Zavar Reza J. Rescue of Dystrophin Protein Expression by Interfering of mRNA Processing. JSSU 2026; 34 (3) :10011-10023
URL: http://jssu.ssu.ac.ir/article-1-6326-fa.html
URL: http://jssu.ssu.ac.ir/article-1-6326-fa.html
مجرد مجید، اصلاحی عطیه، تنی پور حسین، زواررضا جواد. حذف بیان پروتئین دیستروفین با استفاده از تداخل در پردازش mRNA. مجله علمي پژوهشي دانشگاه علوم پزشكي شهید صدوقی يزد. 1405; 34 (3) :10011-10023



متن کامل [PDF 665 kb]
(133 دریافت)
| چکیده (HTML) (235 مشاهده)
References:
1. Kesari A, Pirra LN, Bremadesam L, McIntyre O, Gordon E, Dubrovsky AL, et al. Integrated DNA, cDNA, and protein studies in Becker muscular dystrophy show high exception to the reading frame rule. Hum mutat 2008; 29(5): 728-37.
2. Elangkovan N, Dickson G. Gene Therapy for Duchenne Muscular Dystrophy.J Neuromuscul Dis 2021; 8(s2): S303-S316.
3. Tominari T, Aoki Y. Clinical Development of Novel Therapies for Duchenne Muscular Dystrophy—Current and Future. Neurology and Clinical Neuroscience 2022;11(3): 111-18.
4. Gaedigk R, Law DJ, Fitzgerald-Gustafson KM, McNulty SG, Nsumu NN, Modrcin AC, et al. Improvement in Survival and Muscle Function in an Mdx/Utrn(-/-) Double Mutant Mouse Using A Human Retinal Dystrophin Transgene. Neuromuscul Disord 2006;16(3):192-203.
5. Kawai M. Establishment of an Evaluation Method for Muscular Dystrophy and a Patient Registration System for Clinical Trials. Rinsho shinkeigaku 2009; 49(11): 863-6.
6. Tedesco FS. Human Artificial Chromosomes for Duchenne Muscular Dystrophy and Beyond: Challenges and Hopes. Chromosome Res 2015; 23(1): 135-41.
7. Świątkowska‐Flis B, Zdolińska‐Malinowska I, Sługocka D, Boruczkowski D. The Use of Umbilical Cord‐Derived Mesenchymal Stem Cells in Patients with Muscular Dystrophies: Results from Compassionate Use in Real‐Life Settings. Stem Cells Transl Med 2021; 10(10): 1372-83.
8. Koutsoulidou A, Koutalianos D, Georgiou K, Kakouri A, Oulas A, Tomazou M, et al. Serum Mirnas as Biomarkers for the Rare Types of Muscular Dystrophy. Neuromuscular Disorders 2022; 32(4): 332-46.
9. Przymuszała PM, Martyniak A, Kwiatkowska J, Meyer Szary M, Śledzińska K, Wierzba J, et al. Generation of Human Induced Pluripotent Stem Cell Line Derived from Becker Muscular Dystrophy Patient with CRISPR/Cas9-Mediated Correction of DMD Gene Mutation.Stem Cell Res 2024; 76: 103327.
10. Matsuo M, Takeshima Y. Rescue of Dystrophin Mrna of Duchenne Muscular Dystrophy by Inducing Exon Skipping. Acta Myol 2005; 24(2): 110-4.
11. Brolin C, Shiraishi T. Antisense Mediated Exon Skipping Therapy for Duchenne Muscular Dystrophy (DMD). Artif DNA PNA XNA 2011; 2(1): 6-15.
12. Cirak S, Arechavala-Gomeza V, Guglieri M, Feng L, Torelli S, Anthony K, et al. Exon Skipping and Dystrophin Restoration in Patients with Duchenne Muscular Dystrophy after Systemic Phosphorodiamidate Morpholino Oligomer Treatment: An Open-Label, Phase 2, Dose-Escalation Study. Lancet 2011; 378(9791): 595-605.
13. Nakano S, Ozasa S, Yoshioka K, Fujii I, Mitsui K, Nomura K, et al. Exon-Skipping Events in Candidates for Clinical Trials of Morpholino. Pediatr Int 2011; 53(4): 524-9.
14. Hoogaars WM, Mouisel E, Pasternack A, Hulmi JJ, Relizani K, Schuelke M, et al. Combined Effect of AAV-U7-Induced Dystrophin Exon Skipping and Soluble Activin Type IIB Receptor in Mdx Mice. Human Gene Ther 2012; 23(12): 1269-79.
15. Le Hir M, Goyenvalle A, Peccate C, Precigout G, Davies KE, Voit T, et al. AAV Genome Loss From Dystrophic Mouse Muscles During AAV-U7 snRNA-mediated Exon-skipping Therapy. Mol Ther 2013; 21(8): 1551-558.
16. Hart CC, Lee YI, Xie J, Gao G, Lin BL, Hammers DW, et al. Potential Limitations of Microdystrophin Gene Therapy for Duchenne Muscular Dystrophy. JCI Insight 2024; 9(11): e165869.
17. Mendell J, Muntoni F, McDonald C, Mercuri E, Ciafaloni E, et al. AAV Gene Therapy for Duchenne Muscular Dystrophy: The EMBARK Phase 3 Randomized Trial. Nat Med 2025; 31(1): 332-41.
18. Chwalenia K, Feng V, Hemmer N, Friedrichsen H, Vorobieva L, et al. AAV Microdystrophin Gene Replacement Therapy for Duchenne Muscular Dystrophy: Progress and Prospects. Gene Ther 2025; 32(5): 447-61.
19. Chicoine LG, Rodino-Klapac LR, Shao G, Xu R, Bremer WG, Camboni M, et al. Vascular Delivery of Raavrh74.MCK.GALGT2 to the Gastrocnemius Muscle of the Rhesus Macaque Stimulates the Expression of Dystrophin and Laminin Α2 Surrogates. Mol Ther 2014; 22(4): 713-24.
20. Salva MZ, Himeda CL, Tai PW, Nishiuchi E, Gregorevic P, et al. Design of Tissue-Specific Regulatory Cassettes for High-Level Raav-Mediated Expression in Skeletal and Cardiac Muscle. Mol Ther 2007; 15(2): 320-9.
21. Niks EH, Aartsma-Rus A. Exon Skipping: A First in Class Strategy for Duchenne Muscular Dystrophy. Expert Opin Biol Ther 2017; 17(2): 225-36.
22. van Vliet L, de Winter CL, van Deutekom JC, van Ommen GJ, Aartsma-Rus A. Assessment of the Feasibility of Exon 45-55 Multiexon Skipping for Duchenne Muscular Dystrophy. BMC Med Gen 2008; 9: 105.
23. Aslanidis C, de Jong PJ. Ligation-Independent Cloning of PCR Products (LIC-PCR). Nucleic Acids Res 1990; 18(20): 6069-74.
24. Wendrich JR, Liao CY, van den Berg WA, De Rybel B, Weijers D. Ligation-Independent Cloning For Plant Research. Methods Mol Biol 2015; 1284: 421-31.
25. Schmid-Burgk JL, Schmidt T, Hornung V. Ligation-Independent Cloning (LIC) Assembly of TALEN Genes. Methods Mol Biol 2015; 1239: 161-9.
26. Noori-Daloii MR, Mojarrad M, Rashidi-Nezhad A, Kheirollahi M, Shahbazi A, Khaksari M, et al. Use of Sirna in Knocking Down of Dopamine Receptors, A Possible Therapeutic Option in Neuropsychiatric Disorders. Mol biol Rep 2012; 39(2): 2003-10.
27. Nesmith AP, Wagner MA, Pasqualini FS, O'Connor BB, Pincus MJ, August PR, et al. A Human in Vitro Model of Duchenne Muscular Dystrophy Muscle Formation and Contractility. J Cell Biol 2016; 215(1): 47-56.
متن کامل: (43 مشاهده)
مقدمه
دیستروفی عضلانی دوشن (DMD) یکی از شایعترین اختلالات ژنتیکی است که از هر 3500 تولد مرد 1 نفر اتفاق میافتد. DMD با تخریب شدید پیشرونده عضلانی مشخص میشود که منجر به آتروفی و ضعف عضلات اسکلتی می شود. پیشرفت بیماری منجر به از دست دادن تحرک و محدود شدن ویلچر بیمار تا سن 15 سالگی میشود. تقریباً همه بیماران DMD تا سن 30 سالگی میمیرند (1). تا به امروز، چندین راهکار درمانی، مانند انتقال ژن مینی یا میکرودیستروفین، راهکار کریسپر (CRISPER) ،درمان با سلولهای بنیادی، القاء بیان یوتروفین (utrophin expression)و درمان آنتیسنس به منظور بیان عملکردی دیستروفین در سلولهای عضلانی توسعه یافتهاند (9-2). راهکار پرش ژن (gene skipping) یا اگزون (exon skipping) یکی از امیدوارکنندهترین راهکارهای مبتنی بر نجات چارچوب خواندن باز دیستروفین در ژنهای جهشیافته خارج از چارچوب است که منجر به بیان پروتئین دیستروفین کوتاه شده اما عملکردی میشود (9،10). این تکنیک، یک روش ژنتیکیدرمانی است که در آن با تغییر بهویژه فرایند پردازش (RNA splicing) یک اگزون خاص از روی mRNA حذف گردیده و یک پروتئین کوتاهتر ولی دارای عملکرد تولید میشود. در این استراتژی از الیگونوکلئوتیدهای آنتیسنس (AO) استفاده میشود که با توالیهای پیرایشی یا پیرایش توالی تنظیمی اگزونهای خاص تداخل میکنند که منجر به حذف اگزون مورد نظر در سطح pre-mRNA میشود. کاربرد بالینی راهکار پرش اگزون اخیراً در برخی آزمایشات بالینی مستقل با استفاده از الیگونوکلئوتیدهای اصلاح شده شیمیایی که اگزون 51 دیستروفین را هدف قرار میدهند، نشان داده شده است (11،12). استفاده از سیستم تحویل ویروس مرتبط با آدنو (AAV) بریا انتقال وکتورهای بیان کننده الیگونوکلئوتید، نتایج امیدوارکنندهای را برای درمان DMD نشان میدهد. وکتور ویروسی AAV استفاده میکنند تا نسخهای اصلاحشده از ژن دیستروفین را به سلولهای عضلانی منتقل کنند (13،14). پروتئین تولیدی، یک نسخه کوتاهتر ولی کارآمد از ژن دیستروفین است که باعث بهبود عملکرد عضلات و کند شدن روند بیماری میشود (17-15). آقای Chicoine L. و همکاران نشان دادند که افزایش بیان GALGT2 در عضله اسکلتی رزوس میتواند گلیکوزیلاسیون آلفادیستروگلیکانها زا تحزیک نموده و بیان پروتئینهای باند کننده دیستروفین سیناپسی را افزایش دهد که میتوانند در درمان برخی اشکال دیستروفی عضلانی بهکار روند (18). در سال 2007 گروه Salvaاز ویروس AVV برای انتقال ژن دیستروفین به موشهای مدل دیستروفی عضلانی (MDX) استفاده نمودند. آنها نشان دادند که با استفاده از این وکتور بیان دیستروفین در بافت قلب و عضله بسیار بیشتر از سایر بافت است (19). تیم Mendell J. در سال 2025 کاربرد بالقوه درمانی ونیز مشکلات و کاستیها این تکنیک ها را نشان دادند (16) آنها برای انتقال ژن از وکتور AVV Rh74 استفاده نمودند . آنها ورود ویروس به داخل سلول عضله و بیان دیستروفین را نشان دادند. اما به دلیل بروز برخی عوارض ناخواسته مانند مشکلات مربوط به سیستم کمپلمان آنها پیشنهاد کردند که دز ، زمان مطالعه و شاخص های عملکردی بیشتری بررسی گردد (16). تیم Niks و همکاران پیشرفتهای اولیه در کارآزماییهای انسانی مانند استفاده از داروهایی مانند Eteplirsen و Drisapersen را بررسی کردند (20). آنها نشان دادند که این درمانها، هرچند تولید دیستروفین کامل را ممکن نمیکنند، اما سطح قابل اندازهگیری دیستروفین کوتاه ولی دارای عملکرد را افزایش داده و میتوانند روند تحلیل عضلانی را کند کنند. اگر چه اشکالاتی مانند کم بودن دیستروفین بیان شده، نیاز به تزریق پیوسته، ... را ذکر نمودند .آنها استفاده از پرش اگزونی بهعنوان رهیافتی موثر و اختصاصی برای در مان دوشن را پیشنهاد کردند (20). همانگونه که گفته شد با وجود اثرات مثبت این درمان، بازده پایین انتقال رشتههای آنتیسنس به سلولهای عضلانی بهعنوان چالش اصلی در استفاده از این استراتژی باقی مانده است. بهعلاوه ناهمگونی آللی زیاد دیستروفی عضلانی دوشن (DMD) باعث میشود که انواع الیگونوکلئوتیدهای آنتیسنس مختلف برای درمان بیماران مورد نیاز باشد (15) که یک مشکل چالش برانگیز برای استفاده از وکتورهای AAV در این راهکار درمانی است. بنابراین هدف این مطالعه، ساخت وکتوری با استفاده از روش شبیهسازی مستقل از اتصال (Ligation-independent cloning) یا LIC است که بهوسیله آن بتوان چندین کاست مستقل را در یک وکتور منفرد شبیهسازی کرد. در این مطالعه از شبیهسازی مستقل از لیگیشن (Ligation independent cloning) استفاده گردید که میتوان از آن برای شبیهسازی چندین قطعه انتقالی مستقل بدون نیاز به هضم اندونوکلئازی محدودالاثراستفاده گردید (18-16). در مطالعه حاضر، یک وکتور AAV حامل یک کاست اصلاحشده RNA هستهای کوچک U7(U7snRNA) تولید گردید که اگزون 50 یا 51 الیگونوکلئوتید آنتیسنس را با استفاده از شبیهسازی مستقل از لیگیشن (LIC) بیان میکند. نتایج نشان دهنده کاربرد LIC در تولید بردارهای AAV چند کاست است.
روش بررسی
مواد: تمام پرایمرهای مورد استفاده در این مطالعه در جدول شماره 1 لیست شده است. DNA ژنومیک موش از سویه Balb/c بهدست آمده است و تمام طراحیهای پرایمرها بر اساس ژنوم رفرنس این سویه انجام گرفته است.
حذف کاست بیان ژن نوترکیب از پلاسمید pAAV-MCS: برای تولید پلاسمید پایه اولیه برای ساخت سازه بیان قطعه نوترکیب از پلاسمید pAAV-MCS به عنوان پایه اولیه استفاده شد. به منظور تولید این پلاسمید توالی حاوی پروموتر CMV، ناحیه MCS و توالی HGH-pA-terminator با استفاده از واکنش inversPCR و پرایمرهای معکوس از پلاسمید حذف شدند. این قطعه محصول PCR پلاسمید pAAV-trunc نامیده شد (19).
تولید پلاسمید AAV حاوی ژن U7 موشی: توالی ژن طبیعی U7 موشی با استفاده از واکنش PCR و پرایمرهای اختصاصی ناحیه بالادست و پایین دست این ژن از DNA ژنومیک موش (Mus Musculus) تکثیر و جدا شد. در مرحله بعد با استفاده از تکنولوژی ligase independent cloning (LIC) توالی ژن U7 طبیعی موشی شامل توالی پروموتر U7، توالی کد کننده U7 و توالی انتهایی 3’ ژن U7 به داخل پلاسمید AAV حاصل از مرحله قبل وارد شد. پلاسمید حاصله با استفاده از تعیین توالی تایید ماهیت شد و به عنوان pAAV-U7 نامیده شد.
تولید پلاسمید AAV حاوی توالی U7 فاقد دومین اتصال به DNA: با استفاده از واکنش invers PCR پلاسمید دارای حذف ناحیه دومین اتصال به DNA توالی U7 تکثیر شد و توالی pAAV-U7del نامیده شد (جدول 1).
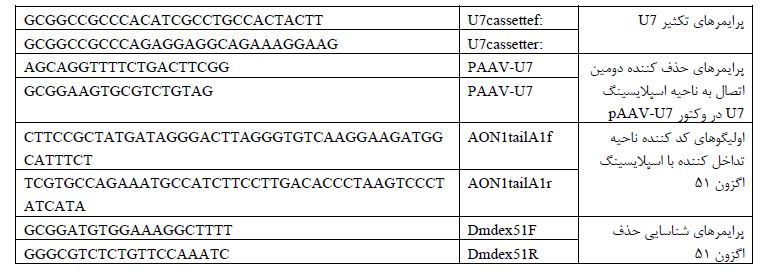
جدول 1: پرایمرهای مورد استفاده در فرایند تولید پلاسمید بیان کننده توالی تداخل کننده با اسپلایسینگ اگزون 51 و بررسی حذف اگزون 51 با استفاده از RT-PCR
تولید پلاسمید AAV حاوی کاستU7-exon skipping (ES)ex51-tail: توالی هیبرید شامل توالی مکمل ناحیه exonic splicing enhancer (ESE) اگزون 51 و توالی دارای ناحیه اتصال با تمایل بالا به hnRNPA1 (بنام tailA1) بهصورت سنتتیک ساخته شد. این توالی طوری طراحی شده بود که دارای همپوشانی با توالیهای دو انتهای pAAV-U7del در دو انتهای خود بود. این دو توالی با استفاده از تکنولوژی LIC با یکدیگر تلفیق شده و تولید پلاسمید pAAV-U7ESex51tail را دادند. این پلاسمید به داخل باکتری DH5Alpha ترانسفورم و تکثیر شدند. ماهیت توالی این پلاسمید با استفاده از تعیین توالی تایید شد.
تولید ویروس AAVES51: ویروس بیان کننده توالی U7-ESex51tail با کوترانسفکشن پلاسمید pAAV-U7ESex51tail و پلاسمیدهای کمکی pAAV-Rec و pHelper (که حاوی ژنهای cap ، rep و دیگر ژنهای مورد نیاز برای تولید ویروس هستند) تولید شدند. برای این منظور سلولهای HEK293 کشت داده شده و با استفاده از ماده Lipofectamin 2000 ترانسفکت شدند. سلولها 48 ساعت پس از ترانسفکشن لیز شده و ذرات ویروسی با استفاده از گرادیان سزیم کلرید و اولتراسانتریفوژ تخلیص و تغلیظ شدند.
کشت سلولهای میوبلاست و آلوده سازی با ویروس: سلولهای میوبلاست انسانی نامیرا شده در محیط کشت RPMI1640 حاوی 20% سرم جنینی گاو( FBS)، و آنتیبیوتیک پنیسیلین/استرپتومایسین کشت داده شدند. تعداد 105 سلول در هر ول از پلیت 12 خانه کشت داده شدند و پس از 24 ساعت تعداد 106 پارتیکل ویروسی به محیط سلولها اضافه شد. بعد از گذشت 4 ساعت محیط سلولها عوض شده و پس از 48 ساعت سلولها برای بررسی میزان بیان RNA و پروتئین دیستروفین مورد استفاده قرار گرفتند (20).
استخراج RNA و واکنش RT-PCR: از سلولهای آلوده شده با ویروس AAVES51، RNA استخراج گردید. برای این منظور از محلول استخراج RNA (Tripure) ساخت کمپانی Roche استفاده شد. مراحل استخراج دقیقا بر طبق دستورالعمل کیت انجام گرفت. در مرحله بعد با استفاده از کیت سنتز cDNA aquapower ساخت شرکت بیونیر و بر اساس دستورالعمل شرکت سازنده cDNA ساخته شد. روش انجام کار به اختصار بهصورت زیر بود: برای ساخت cDNA از µg 1 RNA بهعنوان الگو استفاده شد. این میزان RNA به همراه 1 میکرولیتر پرایمر رندوم هگزامر به لوله حاوی پرمیکس لیوفلیزه RT افزوده و با استفاده از آب مقطر به حجم 20 میکرولیتر رسانیده شد. در نهایت محلول واکنش به مدت 10 دقیقه در 25 درجه، 60 دقیقه در دمای 42 درجه سانتیگراد و 5 دقیقه در دمای 90 درجه سانتیگراد انکوبه شد. به منظور بررسی کیفیت نمونه cDNA ساخته شده در واکنش قبلی، با استفاده از این نمونه و پرایمرهای اختصاصی ژن HPRT یک واکنش PCR انجام گردید. پس از تایید کیفیت cDNA تولید شده، با استفاده از پرایمرهای اختصاصی طراحی شده برای ناحیه بالادست و پایین دست اگزون 51 ژن دیستروفین واکنش PCR انجام شد. این پرایمرها طوری طراحی شده بودند که وجود یا عدم وجود اگزون 51 با بررسی طول محصول PCR مشخص شود. در واقع پرایمر فوروارد در انتهای 3’ اگزون 50 و پرایمر فوروارد در انتهای 5’ اگزون 52 قرار گرفته بود. در صورت حذف اگزون 51 طول محصول PCR با این پرایمرها 135جفت باز و در صورت حضور اگزون 51 این محصول 368 جفت باز طول خواهد داشت.
شرایط واکنش PCR وسترن بلات: پروتئین کامل سلول از فاز ارگانیک بهدست آمده از مراحل استخراج RNA تخلیص شد. برای این منظور به میزان 4 برابر حجم محلول ارگانیک، به این محلول استون سرد اضافه شد و این محلول به مدت 30 دقیقه در دمای منفی 20 درجه انکوبه شد. سپس این محلول به مدت 10 دقیقه و با حداکثر سرعت سانتریفوژ شد. محلول رویی دور ریخته شد و رسوب حاصله در مجاورت هوا خشک شد. پروتئین استخراج شده با 4X SDS sample beffer مخلوط شد و به این مخلوط آب مقطر اضافه شد تا غلظت نهایی بافر به 1X برسد. حجمی از این نمونه که حاوی 20 میکروگرم از پروتئین استخراج شده بود، بر روی ژل اکریل آمید (12% resolving gel, 5% stacking gel)، لود شد و با ولتاژ 200 به مدت 2 ساعت ران شد. در مرحله بعد پروتئین ها به ممبران (PVDF) منتقل شدند (این عمل با استفاده از ولتاژ 200 به مدت 2 ساعت انجام شد). ممبران به مدت 2 ساعت همراه با shacking با محلول بلاکینگ (BSA 1% در محلول TBST) انکوبه شد. در مرحله بعد به مدت 1 ساعت ممبران با محلول آنتیبادی اولیه (رقت 1:100 در بافر بلاکینگ) انکوبه شد و پس از شستشوی ممبران با محلول 1X TBST (سه بار هر بار 5 دقیقه همراه با shacking) به مدت 1 ساعت با آنتیبادی ثانویه (رقت 1:2500 در بافر بلوککننده) همانند شرایط قبلی انکوبه شد. پس از 3 بار شستشو با 1X TBST، ممبران به مدت 5 الی 15 دقیقه با محلول سوبسترای رنگزا (محلول DAB حاوی H2O2 3%) انکوبه شد تا باندهای پروتئین رویت شوند. پس از مرئی شدن باندهای پروتئین، ممبران با آب مقطر شستشو داده شد و از ممبران عکسبرداری شد. با استفاده از نرمافزار دانسیتومتری میزان دانسیته باندهای مشاهده شده، اندازه گیری شدند.
نتایج
حذف موفق کاست بیان ژن نوترکیب از پلاسمید pAAV-MCS: بهمنظور تولید یک پلاسمید پایه AAV فاقد عناصر بیان ژن خارجی، واکنش inverse PCR بر روی پلاسمید pAAV-MCS انجام شد. نتایج الکتروفورز ژل آگارز محصول PCR نشان داد که قطعه موردنظر با اندازه پیشبینیشده با موفقیت تکثیر شده است (شکل 1). حذف توالیهای پروموتر CMV، ناحیه MCS و توالی خاتمهدهنده HGH-pA بهطور کامل انجام شده و هیچ باند اضافی یا نشانهای از تکثیر ناقص مشاهده نشد.
پلاسمید حاصل که با نام pAAV-trunc شناخته شد، بهعنوان اسکلت پایه برای ورود توالیهای U7 در مراحل بعدی مورد استفاده قرار گرفت. حذف این کاست بیان از اهمیت بالایی برخوردار است، زیرا امکان کنترل دقیقتر بیان RNAهای کوچک غیرکدکننده نظیر U7 را فراهم کرده و از تداخل پروموترهای قوی ویروسی با عملکرد طبیعی سیستم اسپلایسینگ جلوگیری میکند (شکل 1 الف).
تولید و شناسایی پلاسمید pAAV-U7 حاوی ژن طبیعی U7 موشی: ژن U7 موشی با استفاده از پرایمرهای اختصاصی از DNA ژنومیک موش با موفقیت تکثیر شد. بررسی محصول PCR نشاندهنده وجود یک باند یکتا با اندازه مورد انتظار بود که بیانگر اختصاصیت بالای پرایمرها و صحت تکثیر ژن هدف است (شکل 1 ب). در مرحله بعد، توالی U7 شامل پروموتر طبیعی، ناحیه کدکننده و توالی انتهایی 3´ به داخل پلاسمید pAAV-trunc بهکمک روش ligase independent cloning (LIC) وارد شد. تعیین توالی پلاسمید نوترکیب نشان داد که توالی U7 بدون هیچگونه حذف، اضافه یا جهش نقطهای در محل صحیح قرار گرفته است. این سازه با نام pAAV-U7 شناسایی شد و بهعنوان مرجع عملکرد طبیعی RNA U7 در ادامه مطالعه مورد استفاده قرار گرفت.
حذف هدفمند دومین اتصال به DNA در پلاسمید pAAV-U7del: برای بررسی نقش دومین اتصال به DNA در عملکرد RNA U7، حذف هدفمند این ناحیه با استفاده از inverse PCR انجام شد. نتایج الکتروفورز محصول PCR نشان داد که پلاسمید حذف شده دارای اندازه کوچکتر نسبت به پلاسمید pAAV-U7 اولیه است که حذف موفق ناحیه هدف را تأیید میکند (شکل 3). نتایج تعیین توالی نیز نشان داد که حذف دومین اتصال به DNA بدون تأثیر بر سایر نواحی ساختاری U7 انجام شده است. پلاسمید حاصل با نام pAAV-U7del بهعنوان بستر مناسب برای ورود توالیهای exon skipping طراحیشده مورد استفاده قرار گرفت.
ساخت و تأیید پلاسمید:pAAV-U7ESex51tail توالی هیبرید سنتتیک شامل ناحیه مکمل ESE اگزون 51 ژن دیستروفین و توالی tailA1 با تمایل اتصال بالا به hnRNPA1 طراحی و سنتز شد. این توالی بهگونهای طراحی شده بود که از طریق همپوشانی انتهایی، امکان اتصال مستقیم به پلاسمید pAAV-U7del را فراهم کند. نتایج کلونینگ با استفاده از روش LIC نشان داد که توالی هیبرید با موفقیت در محل مناسب وارد پلاسمید شده است. بررسی تکثیر PCR و الکتروفورز نشان داد که سازه نهایی کاملاً مطابق طراحی اولیه است (شکل 1ج). پلاسمید حاصل با نام pAAV-U7ESex51tail برای تولید ویروس نوترکیب مورد استفاده قرار گرفت.
تولید و تخلیص ویروس نوترکیبAAVES51: ویروس نوترکیب AAVES51 با کوترانسفکشن پلاسمید pAAV-U7ESex51tail به همراه پلاسمیدهای کمکی در سلولهای HEK293 تولید شد. بررسی مورفولوژی سلولها پس از ترانسفکشن نشاندهنده سلامت نسبی سلولها و عدم بروز سمیت شدید بود. پس از تخلیص ویروس با استفاده از گرادیان سزیم کلرید، ذرات ویروسی با خلوص مناسب برای آلودهسازی سلولی بهدست آمدند.
آلودهسازی سلولهای میوبلاست انسانی با ویروس AAVES51: سلولهای میوبلاست انسانی نامیرا شده با ویروس AAVES51 آلوده شدند. بررسی مورفولوژی سلولی پس از 48 ساعت نشان داد که سلولها زندهمانی بالایی داشته و تغییرات مورفولوژیک قابلتوجهی مشاهده نشد (شکل 2). این یافتهها نشان میدهد که ویروس AAVES51 در شرایط آزمایشگاهی مورد استفاده، اثر سمی قابلتوجهی بر سلولهای میوبلاست انسانی ندارد و برای مطالعات عملکردی مناسب است.
القای حذف اگزون 51 در سطحRNA: نتایج RT-PCR با استفاده از پرایمرهای اختصاصی اطراف اگزون 51 ژن دیستروفین نشان داد که در سلولهای آلودهشده با ویروس AAVES51، باند غالب با طول 210 جفت باز مشاهده شد که بیانگر حذف موفق اگزون 51 است (شکل 3). در مقابل، در نمونههای کنترل غیرآلوده، باند 450 جفت بازی مربوط به حضور اگزون 51 مشاهده شد. این تفاوت آشکار در الگوی باندها نشاندهنده کارایی بالای سیستم U7-ESex51-tail در القای exon skipping هدفمند میباشد.
افزایش بیان پروتئین دیستروفین در سطح پروتئین: نتایج وسترن بلات نشان داد که در سلولهای تیمار شده با ویروس AAVES51، شدت باند پروتئین دیستروفین نسبت به نمونههای کنترل افزایش یافته است (شکل 4). آنالیز دانسیتومتری باندها نیز افزایش قابلتوجه میزان بیان پروتئین دیستروفین را نشان داد. این نتایج بیانگر آن است که حذف اگزون 51 در سطح RNA منجر به بازسازی چهارچوب خوانش ژن دیستروفین و افزایش تولید پروتئین در سطح سلولی شده است.
بررسی حذف اگزون 51 بدنبال آلوده سازی سلول میوبلاست طبیعی با ویروس AAV-ESex51 با استفاده از RT-PCR: آلودهسازی سلولهای میوبلاست طبیعی با ویروس AAV-ESex51 منجر به حذف اگزون 51 از mRNA بالغ دیستروفین شد. بر اساس نتایج بهدست آمده از واکنش RT-PCR بخش قابل توجهی از mRNA استخراج شده از سلول آلوده در مقایسه با سلول کنترل دچار حذف اگزون 51 شده است (شکل3).
***توجه به این نکته ضروری است که بخشی از باند محصول تکثیر توالی واجد اگزون 51 در این واکنش میتواند به علت تکثیر توالیهای hnRNA (نسخه های RNA نابالغ که هنوز تحت فرایند پردازش قرار نگرفتهاند) باشد که نتیجتا راندمان فرایند exon skipping ناشی از آلودهسازی سلول با ویروس AAV-ESex51 احتمالاً بیشتر از آنچه در عکس دیده شده است میباشد.
بررسی حذف اگزون 51 بهدنبال آلودهسازی سلول میوبلاست طبیعی با ویروس AAV-ESex51 با استفاده از وسترن بلات
آلودهسازی سلولهای میوبلاست طبیعی با ویروس AAV-ESex51 منجر به حذف اگزون 51 از mRNA بالغ دیستروفین میشود. این واقعه باعث وقوع جهش تغییر چهارچوب (frameshift) میشود که نهایتا منجر به تخریب RNA و عدم تولید پروتئین دیستروفین میشود (شکل 3).
نتایج حاصل از بررسی وسترن بلات نیز بهطور واضح نشاندهنده کاهش چشمگیر میزان پروتئین دیستروفین در سلولهای آلوده به ویروس میباشد.
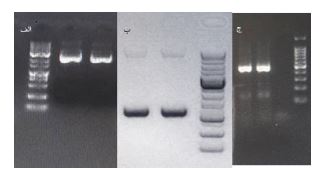
شکل 1: نتایج مراحل مختلف تولید AAV تولید کننده توالی U7-ESex51-tail
الف) الکتروفورز ژل آگارز محصول inverse PCR برای حذف کاست بیان ژن نوترکیب از پلاسمید pAAV-MCS و تولید پلاسمید pAAV-trunc نتیجه این واکنش یک باند حدودا 5 کیلوبازی بود که همانطور که در تصویر دیده میشود تکثیر شده بود. ب) تکثیر ژن U7 موشی از DNA ژنومیک موش و بررسی محصول PCR بر روی ژل آگارز. ج) تأیید ورود توالی U7-ESex51-tail به پلاسمید pAAV-U7del با استفاده از PCR الکتروفورز.

شکل 2: بررسی مورفولوژی و آلودهسازی سلولهای میوبلاست انسانی پس از تیمار با ویروس
AAVES51 تصاویر فلورسانس (ستون چپ) نشاندهنده حضور سلولهای آلودهشده با ویروس میباشند، در حالیکه تصاویر میدان روشن (ستون راست) مورفولوژی طبیعی و تراکم مناسب سلولها را پس از تیمار نشان میدهند. عدم مشاهده تغییرات مورفولوژیک بارز بین نمونههای تیمار شده و کنترل بیانگر عدم سمیت قابلتوجه ویروس AAVES51 در شرایط آزمایشگاهی است. (نوار مقیاس: 100 میکرومتر)
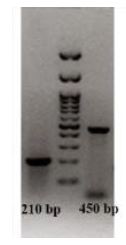
شکل 3: نتایج RT-PCR :بررسی حذف اگزون 51 ژن دیستروفین در سلولهای میوبلاست انسانی پس از آلودهسازی با ویروس AAVES51
باند با اندازه تقریبی 210 جفت باز نشاندهنده حذف اگزون 51 در سلولهای تیمار شده است، در حالیکه باند حدودا 450 جفت بازی در نمونه کنترل بیانگر حضور اگزون 51 میباشد. مارکر وزن مولکولی DNA برای تخمین اندازه محصولات PCR استفاده شده است.
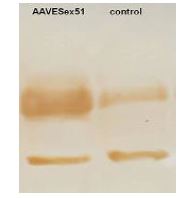
شکل 4: نتایج وسترن بلات بررسی بیان پروتئین دیستروفین در سلولهای میوبلاست DMD آلوده شده با ویروس AAVeESex51 در مقابل سلولهای کنترل.
پس از آلودهسازی با ویروس AAVESex51 افزایش شدت باند مربوط به پروتئین دیستروفین (باند بالایی) در نمونه تیمار شده با AAVESex51 نسبت به نمونه کنترل مشاهده میشود. باند پایینی مربوط به ژن خانهدار بهعنوان کنترل داخلی برای نرمالسازی میزان پروتئین بهکار رفته است.
بحث
بیماری دیستروفی عضلانی دوشن (Duchenne Muscular Dystrophy; DMD) یکی از شایعترین و شدیدترین بیماریهای نوروموسکولار وابسته به جنس است که در اثر جهشهای ایجادکننده فریمشیفت در ژن دیستروفین ایجاد میشود (1). از اواسط دهه ۱۹۹۰ میلادی، معرفی مفهوم exon skipping بهعنوان یک راهبرد درمانی مولکولی، افق تازهای را در درمان این بیماری گشود. اساس این رویکرد بر حذف هدفمند یک یا چند اگزون از پیش mRNA استوار است تا چهارچوب خوانش ژن بازسازی شده و یک پروتئین دیستروفین کوتاهتر اما تا حدی عملکردی تولید شود؛ الگویی که از فرم خفیفتر بیماری یعنی دیستروفی عضلانی بیکر(Becker muscular destrophy ) الهام گرفته شده است. ایده استفاده از پدیده پرش اگزونی (Exon Skipping) بهعنوان یک راهبرد درمانی برای دیستروفی عضلانی دوشن (DMD) از اواسط دهه 1990 میلادی مطرح شد و بهسرعت بهعنوان یکی از واقعگرایانهترین رویکردهای درمانی برای این بیماری شناخته شد. منطق زیربنایی این استراتژی بر این اصل استوار است که حذف هدفمند یک یا چند اگزون میتواند چهارچوب خوانش ژن دیستروفین را بازسازی کرده و منجر به تولید یک پروتئین کوتاهشده اما عملکردی، مشابه آنچه در بیماران مبتلا به دیستروفی بکر مشاهده میشود، گردد(9-2). یکی از مهمترین مزایای استراتژی exon skipping در مقایسه با رویکردهای مبتنی بر انتقال DNA نوترکیب، عدم دستکاری مستقیم ژنوم میزبان است. این ویژگی، خطرات بالقوهای نظیر درج تصادفی توالیهای خارجی در ژنوم، فعالسازی انکوژنها یا اختلال در ژنهای حیاتی را بهطور قابلتوجهی کاهش میدهد. به همین دلیل، exon skipping از همان ابتدا بهعنوان رویکردی ایمنتر نسبت به ژندرمانی کلاسیک مطرح شد. در یک دهه گذشته، مطالعات متعددی کارایی و ایمنی این تکنولوژی را در مدلهای سلولی، حیوانی و حتی کارآزماییهای بالینی نشان دادهاند. استفاده از الیگونوکلئوتیدهای آنتیسنس (AONs) مانند eteplirsen که حذف اگزون 51 را القا میکند، به تأیید سازمان FDA نیز رسیده و شواهد بالینی از افزایش نسبی دیستروفین و کند شدن سیر پیشرفت بیماری ارائه داده است (12-11). با این حال، محدودیت اصلی AONهای سنتتیک، نیمهعمر کوتاه، نیاز به تزریقهای مکرر و نفوذ ناکافی به بافت عضله اسکلتی بوده است. برای غلبه بر این محدودیتها، استفاده از ناقلهای ویروسی بهویژه ویروسهای آدنو-مرتبط (AAV) بهعنوان سیستمهای تحویل پایدار مورد توجه قرار گرفت (20).AAVها به دلیل تمایل بافتی بالا به عضله، ایمنی نسبی و توانایی ایجاد بیان پایدارRNA، گزینهای مناسب برای انتقال کاستهای القاکننده exon skipping محسوب میشوند. مطالعات متعددی نشان دادهاند که بیان پایدار RNAهای مبتنی بر U7 snRNA از طریق AAV میتواند منجر به القای طولانیمدت exon skipping در مدلهای mdx شود. در مطالعه حاضر، ما موفق به طراحی و تولید یک وکتور AAV مبتنی بر U7 اصلاحشده شدیم که حذف هدفمند اگزون 51 ژن دیستروفین را القا میکند. نتایج RT-PCR نشان داد که بخش قابلتوجهی از ترانسکریپتهای تولیدشده در سلولهای میوبلاست آلودهشده، فاقد اگزون 51 هستند. این یافته با نتایج مطالعات پیشین که از سیستم U7 برای القای exon skipping استفاده کردهاند، همخوانی دارد و نشاندهنده کارایی مناسب این سیستم در سطح RNA است. از منظر پروتئینی، نتایج وسترن بلات افزایش بیان دیستروفین در سلولهای تیمار شده را نشان داد. این موضوع از نظر بیولوژیکی حائز اهمیت است، زیرا نشان میدهد که حذف اگزون 51 منجر به بازسازی چهارچوب خوانش و ترجمه موفق mRNA اصلاحشده شده است. هرچند میزان دیستروفین تولیدشده به سطح فیزیولوژیک کامل نرسیده، اما شواهد متعددی نشان دادهاند که حتی مقادیر اندک (۵–15%) دیستروفین میتواند اثرات درمانی قابلتوجهی در بیماران DMD داشته باشد. یکی از دستاوردهای مهم این مطالعه، بومیسازی فناوری تولید ناقلهای AAV القاکننده exon skipping در قالب یک پلتفرم انعطافپذیر است. استفاده از تکنولوژی ligase independent cloning (LIC) برای اولین بار در تولید وکتور AAV-U7 در این مطالعه، امکان طراحی سریع، دقیق و کمهزینه وکتورهای متنوع را فراهم کرده است. این موضوع از منظر پزشکی شخصی(personalized medicine) اهمیت ویژهای دارد، زیرا ژن دیستروفین دارای طیف وسیعی از جهشهای حذفی و مضاعفشدگی است و درمان همه بیماران با یک استراتژی واحد امکانپذیر نیست. پلاسمید پایه pAAV-U7del که در این مطالعه طراحی شد، این قابلیت را دارد که بهعنوان یک اسکلت عمومی برای درج سریع توالیهای exon skipping هدفگیرنده اگزونهای مختلف ژن دیستروفین مورد استفاده قرار گیرد. این ویژگی، زمان تولید وکتورهای جدید را به کمتر از یک ماه کاهش داده و هزینه و پیچیدگی فرایند را بهطور چشمگیری کم میکند. علاوه بر این، امکان طراحی وکتورهایی با چندین کاست U7 مستقل وجود دارد که میتواند حذف همزمان چند اگزون را هدف قرار دهد؛ راهبردی که برای بیماران دارای جهشهای متنوع در نواحی hotspot ژن دیستروفین بسیار جذاب است. با وجود مزایای قابلتوجه exon skipping، این رویکرد محدودیتهایی نیز دارد. مهمترین محدودیت آن، عدم اصلاح دائمی ژنوم است؛ بهطوریکه اثر درمانی وابسته به تداوم بیان RNA القاکننده exon skipping باقی میماند. در این زمینه، فناوریهای ویرایش ژن نظیر CRISPR/Cas9 بهعنوان جایگزینهای بالقوه مطرح شدهان CRISPR میتواند با حذف دائمی اگزونهای معیوب یا اصلاح جهشها، درمانی ریشهایتر ارائه دهد. با این حال، سیستم CRISPR نیز با چالشهای مهمی مواجه است؛ از جمله خطر برشهای خارج از هدف (off-target effects)، پاسخهای ایمنی علیه Cas9، و پیچیدگی کنترل دقیق ویرایش در بافتهای مختلف. در مقابل، exon skipping مبتنی بر RNA رویکردی ایمنتر و قابلکنترلتر محسوب میشود که بهویژه در شرایطی که ایمنی اولویت بالاتری دارد، همچنان گزینهای جذاب است. از دیگر محدودیتهای این مطالعه میتوان به استفاده از مدل سلولی بهجای مدل حیوانی، عدم بررسی عملکرد عضلانی و عدم ارزیابی طولانیمدت پایداری بیان دیستروفین اشاره کرد. همچنین، بررسی دقیق میزان exon skipping در سطح کمی (quantitative) و مقایسه با سایر سیستمهای تحویلی میتواند در مطالعات آینده مدنظر قرار گیرد)(16-18).
نتیجهگیری
در مجموع، نتایج این مطالعه نشان میدهد که ترکیب سیستم U7 اصلاحشده با ناقلهای AAV و استفاده از فناوری LIC میتواند یک پلتفرم کارآمد، منعطف و بومی برای توسعه درمانهای exon skipping در بیماری DMD فراهم کند. این رویکرد میتواند بهعنوان پلی میان درمانهای RNA محور و فناوریهای ویرایش ژن، جایگاه ویژهای در آینده درمان دیستروفی عضلانی دوشن داشته باشد.
سپاسگزاری
مطالعه ماحصل طرح تحقیقاتی با شماره 1575 مصوب دانشگاه شهید صدوقی یزد است.
حامی مالی: دانشگاه شهید صدوقی یزد
تعارض در منافع: وجود ندارد.
ملاحظات اخلاقی
پروپوزال این تحقیق توسط دانشگاه علوم پزشکی شهید صدوقی یزد و مشهد تایید شده است.
.مشارکت نویسندگان
دکتر مجید مجرد و دکتر جواد زواررضادر ارائه ایده، و طراحی مطالعه و تجزیه و تحلیل دادهها ، خانم عطیه اصلاحی و حسین تنیپور در انجام کار آزمایشگاهی، جمعآوری دادهها مشارکت داشته و همه نویسندگان در تدوین، ویرایش اولیه و نهایی مقاله و پاسخگویی به سوالات مرتبط با مقاله سهیم هستند.
دیستروفی عضلانی دوشن (DMD) یکی از شایعترین اختلالات ژنتیکی است که از هر 3500 تولد مرد 1 نفر اتفاق میافتد. DMD با تخریب شدید پیشرونده عضلانی مشخص میشود که منجر به آتروفی و ضعف عضلات اسکلتی می شود. پیشرفت بیماری منجر به از دست دادن تحرک و محدود شدن ویلچر بیمار تا سن 15 سالگی میشود. تقریباً همه بیماران DMD تا سن 30 سالگی میمیرند (1). تا به امروز، چندین راهکار درمانی، مانند انتقال ژن مینی یا میکرودیستروفین، راهکار کریسپر (CRISPER) ،درمان با سلولهای بنیادی، القاء بیان یوتروفین (utrophin expression)و درمان آنتیسنس به منظور بیان عملکردی دیستروفین در سلولهای عضلانی توسعه یافتهاند (9-2). راهکار پرش ژن (gene skipping) یا اگزون (exon skipping) یکی از امیدوارکنندهترین راهکارهای مبتنی بر نجات چارچوب خواندن باز دیستروفین در ژنهای جهشیافته خارج از چارچوب است که منجر به بیان پروتئین دیستروفین کوتاه شده اما عملکردی میشود (9،10). این تکنیک، یک روش ژنتیکیدرمانی است که در آن با تغییر بهویژه فرایند پردازش (RNA splicing) یک اگزون خاص از روی mRNA حذف گردیده و یک پروتئین کوتاهتر ولی دارای عملکرد تولید میشود. در این استراتژی از الیگونوکلئوتیدهای آنتیسنس (AO) استفاده میشود که با توالیهای پیرایشی یا پیرایش توالی تنظیمی اگزونهای خاص تداخل میکنند که منجر به حذف اگزون مورد نظر در سطح pre-mRNA میشود. کاربرد بالینی راهکار پرش اگزون اخیراً در برخی آزمایشات بالینی مستقل با استفاده از الیگونوکلئوتیدهای اصلاح شده شیمیایی که اگزون 51 دیستروفین را هدف قرار میدهند، نشان داده شده است (11،12). استفاده از سیستم تحویل ویروس مرتبط با آدنو (AAV) بریا انتقال وکتورهای بیان کننده الیگونوکلئوتید، نتایج امیدوارکنندهای را برای درمان DMD نشان میدهد. وکتور ویروسی AAV استفاده میکنند تا نسخهای اصلاحشده از ژن دیستروفین را به سلولهای عضلانی منتقل کنند (13،14). پروتئین تولیدی، یک نسخه کوتاهتر ولی کارآمد از ژن دیستروفین است که باعث بهبود عملکرد عضلات و کند شدن روند بیماری میشود (17-15). آقای Chicoine L. و همکاران نشان دادند که افزایش بیان GALGT2 در عضله اسکلتی رزوس میتواند گلیکوزیلاسیون آلفادیستروگلیکانها زا تحزیک نموده و بیان پروتئینهای باند کننده دیستروفین سیناپسی را افزایش دهد که میتوانند در درمان برخی اشکال دیستروفی عضلانی بهکار روند (18). در سال 2007 گروه Salvaاز ویروس AVV برای انتقال ژن دیستروفین به موشهای مدل دیستروفی عضلانی (MDX) استفاده نمودند. آنها نشان دادند که با استفاده از این وکتور بیان دیستروفین در بافت قلب و عضله بسیار بیشتر از سایر بافت است (19). تیم Mendell J. در سال 2025 کاربرد بالقوه درمانی ونیز مشکلات و کاستیها این تکنیک ها را نشان دادند (16) آنها برای انتقال ژن از وکتور AVV Rh74 استفاده نمودند . آنها ورود ویروس به داخل سلول عضله و بیان دیستروفین را نشان دادند. اما به دلیل بروز برخی عوارض ناخواسته مانند مشکلات مربوط به سیستم کمپلمان آنها پیشنهاد کردند که دز ، زمان مطالعه و شاخص های عملکردی بیشتری بررسی گردد (16). تیم Niks و همکاران پیشرفتهای اولیه در کارآزماییهای انسانی مانند استفاده از داروهایی مانند Eteplirsen و Drisapersen را بررسی کردند (20). آنها نشان دادند که این درمانها، هرچند تولید دیستروفین کامل را ممکن نمیکنند، اما سطح قابل اندازهگیری دیستروفین کوتاه ولی دارای عملکرد را افزایش داده و میتوانند روند تحلیل عضلانی را کند کنند. اگر چه اشکالاتی مانند کم بودن دیستروفین بیان شده، نیاز به تزریق پیوسته، ... را ذکر نمودند .آنها استفاده از پرش اگزونی بهعنوان رهیافتی موثر و اختصاصی برای در مان دوشن را پیشنهاد کردند (20). همانگونه که گفته شد با وجود اثرات مثبت این درمان، بازده پایین انتقال رشتههای آنتیسنس به سلولهای عضلانی بهعنوان چالش اصلی در استفاده از این استراتژی باقی مانده است. بهعلاوه ناهمگونی آللی زیاد دیستروفی عضلانی دوشن (DMD) باعث میشود که انواع الیگونوکلئوتیدهای آنتیسنس مختلف برای درمان بیماران مورد نیاز باشد (15) که یک مشکل چالش برانگیز برای استفاده از وکتورهای AAV در این راهکار درمانی است. بنابراین هدف این مطالعه، ساخت وکتوری با استفاده از روش شبیهسازی مستقل از اتصال (Ligation-independent cloning) یا LIC است که بهوسیله آن بتوان چندین کاست مستقل را در یک وکتور منفرد شبیهسازی کرد. در این مطالعه از شبیهسازی مستقل از لیگیشن (Ligation independent cloning) استفاده گردید که میتوان از آن برای شبیهسازی چندین قطعه انتقالی مستقل بدون نیاز به هضم اندونوکلئازی محدودالاثراستفاده گردید (18-16). در مطالعه حاضر، یک وکتور AAV حامل یک کاست اصلاحشده RNA هستهای کوچک U7(U7snRNA) تولید گردید که اگزون 50 یا 51 الیگونوکلئوتید آنتیسنس را با استفاده از شبیهسازی مستقل از لیگیشن (LIC) بیان میکند. نتایج نشان دهنده کاربرد LIC در تولید بردارهای AAV چند کاست است.
روش بررسی
مواد: تمام پرایمرهای مورد استفاده در این مطالعه در جدول شماره 1 لیست شده است. DNA ژنومیک موش از سویه Balb/c بهدست آمده است و تمام طراحیهای پرایمرها بر اساس ژنوم رفرنس این سویه انجام گرفته است.
حذف کاست بیان ژن نوترکیب از پلاسمید pAAV-MCS: برای تولید پلاسمید پایه اولیه برای ساخت سازه بیان قطعه نوترکیب از پلاسمید pAAV-MCS به عنوان پایه اولیه استفاده شد. به منظور تولید این پلاسمید توالی حاوی پروموتر CMV، ناحیه MCS و توالی HGH-pA-terminator با استفاده از واکنش inversPCR و پرایمرهای معکوس از پلاسمید حذف شدند. این قطعه محصول PCR پلاسمید pAAV-trunc نامیده شد (19).
تولید پلاسمید AAV حاوی ژن U7 موشی: توالی ژن طبیعی U7 موشی با استفاده از واکنش PCR و پرایمرهای اختصاصی ناحیه بالادست و پایین دست این ژن از DNA ژنومیک موش (Mus Musculus) تکثیر و جدا شد. در مرحله بعد با استفاده از تکنولوژی ligase independent cloning (LIC) توالی ژن U7 طبیعی موشی شامل توالی پروموتر U7، توالی کد کننده U7 و توالی انتهایی 3’ ژن U7 به داخل پلاسمید AAV حاصل از مرحله قبل وارد شد. پلاسمید حاصله با استفاده از تعیین توالی تایید ماهیت شد و به عنوان pAAV-U7 نامیده شد.
تولید پلاسمید AAV حاوی توالی U7 فاقد دومین اتصال به DNA: با استفاده از واکنش invers PCR پلاسمید دارای حذف ناحیه دومین اتصال به DNA توالی U7 تکثیر شد و توالی pAAV-U7del نامیده شد (جدول 1).
جدول 1: پرایمرهای مورد استفاده در فرایند تولید پلاسمید بیان کننده توالی تداخل کننده با اسپلایسینگ اگزون 51 و بررسی حذف اگزون 51 با استفاده از RT-PCR
تولید پلاسمید AAV حاوی کاستU7-exon skipping (ES)ex51-tail: توالی هیبرید شامل توالی مکمل ناحیه exonic splicing enhancer (ESE) اگزون 51 و توالی دارای ناحیه اتصال با تمایل بالا به hnRNPA1 (بنام tailA1) بهصورت سنتتیک ساخته شد. این توالی طوری طراحی شده بود که دارای همپوشانی با توالیهای دو انتهای pAAV-U7del در دو انتهای خود بود. این دو توالی با استفاده از تکنولوژی LIC با یکدیگر تلفیق شده و تولید پلاسمید pAAV-U7ESex51tail را دادند. این پلاسمید به داخل باکتری DH5Alpha ترانسفورم و تکثیر شدند. ماهیت توالی این پلاسمید با استفاده از تعیین توالی تایید شد.
تولید ویروس AAVES51: ویروس بیان کننده توالی U7-ESex51tail با کوترانسفکشن پلاسمید pAAV-U7ESex51tail و پلاسمیدهای کمکی pAAV-Rec و pHelper (که حاوی ژنهای cap ، rep و دیگر ژنهای مورد نیاز برای تولید ویروس هستند) تولید شدند. برای این منظور سلولهای HEK293 کشت داده شده و با استفاده از ماده Lipofectamin 2000 ترانسفکت شدند. سلولها 48 ساعت پس از ترانسفکشن لیز شده و ذرات ویروسی با استفاده از گرادیان سزیم کلرید و اولتراسانتریفوژ تخلیص و تغلیظ شدند.
کشت سلولهای میوبلاست و آلوده سازی با ویروس: سلولهای میوبلاست انسانی نامیرا شده در محیط کشت RPMI1640 حاوی 20% سرم جنینی گاو( FBS)، و آنتیبیوتیک پنیسیلین/استرپتومایسین کشت داده شدند. تعداد 105 سلول در هر ول از پلیت 12 خانه کشت داده شدند و پس از 24 ساعت تعداد 106 پارتیکل ویروسی به محیط سلولها اضافه شد. بعد از گذشت 4 ساعت محیط سلولها عوض شده و پس از 48 ساعت سلولها برای بررسی میزان بیان RNA و پروتئین دیستروفین مورد استفاده قرار گرفتند (20).
استخراج RNA و واکنش RT-PCR: از سلولهای آلوده شده با ویروس AAVES51، RNA استخراج گردید. برای این منظور از محلول استخراج RNA (Tripure) ساخت کمپانی Roche استفاده شد. مراحل استخراج دقیقا بر طبق دستورالعمل کیت انجام گرفت. در مرحله بعد با استفاده از کیت سنتز cDNA aquapower ساخت شرکت بیونیر و بر اساس دستورالعمل شرکت سازنده cDNA ساخته شد. روش انجام کار به اختصار بهصورت زیر بود: برای ساخت cDNA از µg 1 RNA بهعنوان الگو استفاده شد. این میزان RNA به همراه 1 میکرولیتر پرایمر رندوم هگزامر به لوله حاوی پرمیکس لیوفلیزه RT افزوده و با استفاده از آب مقطر به حجم 20 میکرولیتر رسانیده شد. در نهایت محلول واکنش به مدت 10 دقیقه در 25 درجه، 60 دقیقه در دمای 42 درجه سانتیگراد و 5 دقیقه در دمای 90 درجه سانتیگراد انکوبه شد. به منظور بررسی کیفیت نمونه cDNA ساخته شده در واکنش قبلی، با استفاده از این نمونه و پرایمرهای اختصاصی ژن HPRT یک واکنش PCR انجام گردید. پس از تایید کیفیت cDNA تولید شده، با استفاده از پرایمرهای اختصاصی طراحی شده برای ناحیه بالادست و پایین دست اگزون 51 ژن دیستروفین واکنش PCR انجام شد. این پرایمرها طوری طراحی شده بودند که وجود یا عدم وجود اگزون 51 با بررسی طول محصول PCR مشخص شود. در واقع پرایمر فوروارد در انتهای 3’ اگزون 50 و پرایمر فوروارد در انتهای 5’ اگزون 52 قرار گرفته بود. در صورت حذف اگزون 51 طول محصول PCR با این پرایمرها 135جفت باز و در صورت حضور اگزون 51 این محصول 368 جفت باز طول خواهد داشت.
شرایط واکنش PCR وسترن بلات: پروتئین کامل سلول از فاز ارگانیک بهدست آمده از مراحل استخراج RNA تخلیص شد. برای این منظور به میزان 4 برابر حجم محلول ارگانیک، به این محلول استون سرد اضافه شد و این محلول به مدت 30 دقیقه در دمای منفی 20 درجه انکوبه شد. سپس این محلول به مدت 10 دقیقه و با حداکثر سرعت سانتریفوژ شد. محلول رویی دور ریخته شد و رسوب حاصله در مجاورت هوا خشک شد. پروتئین استخراج شده با 4X SDS sample beffer مخلوط شد و به این مخلوط آب مقطر اضافه شد تا غلظت نهایی بافر به 1X برسد. حجمی از این نمونه که حاوی 20 میکروگرم از پروتئین استخراج شده بود، بر روی ژل اکریل آمید (12% resolving gel, 5% stacking gel)، لود شد و با ولتاژ 200 به مدت 2 ساعت ران شد. در مرحله بعد پروتئین ها به ممبران (PVDF) منتقل شدند (این عمل با استفاده از ولتاژ 200 به مدت 2 ساعت انجام شد). ممبران به مدت 2 ساعت همراه با shacking با محلول بلاکینگ (BSA 1% در محلول TBST) انکوبه شد. در مرحله بعد به مدت 1 ساعت ممبران با محلول آنتیبادی اولیه (رقت 1:100 در بافر بلاکینگ) انکوبه شد و پس از شستشوی ممبران با محلول 1X TBST (سه بار هر بار 5 دقیقه همراه با shacking) به مدت 1 ساعت با آنتیبادی ثانویه (رقت 1:2500 در بافر بلوککننده) همانند شرایط قبلی انکوبه شد. پس از 3 بار شستشو با 1X TBST، ممبران به مدت 5 الی 15 دقیقه با محلول سوبسترای رنگزا (محلول DAB حاوی H2O2 3%) انکوبه شد تا باندهای پروتئین رویت شوند. پس از مرئی شدن باندهای پروتئین، ممبران با آب مقطر شستشو داده شد و از ممبران عکسبرداری شد. با استفاده از نرمافزار دانسیتومتری میزان دانسیته باندهای مشاهده شده، اندازه گیری شدند.
نتایج
حذف موفق کاست بیان ژن نوترکیب از پلاسمید pAAV-MCS: بهمنظور تولید یک پلاسمید پایه AAV فاقد عناصر بیان ژن خارجی، واکنش inverse PCR بر روی پلاسمید pAAV-MCS انجام شد. نتایج الکتروفورز ژل آگارز محصول PCR نشان داد که قطعه موردنظر با اندازه پیشبینیشده با موفقیت تکثیر شده است (شکل 1). حذف توالیهای پروموتر CMV، ناحیه MCS و توالی خاتمهدهنده HGH-pA بهطور کامل انجام شده و هیچ باند اضافی یا نشانهای از تکثیر ناقص مشاهده نشد.
پلاسمید حاصل که با نام pAAV-trunc شناخته شد، بهعنوان اسکلت پایه برای ورود توالیهای U7 در مراحل بعدی مورد استفاده قرار گرفت. حذف این کاست بیان از اهمیت بالایی برخوردار است، زیرا امکان کنترل دقیقتر بیان RNAهای کوچک غیرکدکننده نظیر U7 را فراهم کرده و از تداخل پروموترهای قوی ویروسی با عملکرد طبیعی سیستم اسپلایسینگ جلوگیری میکند (شکل 1 الف).
تولید و شناسایی پلاسمید pAAV-U7 حاوی ژن طبیعی U7 موشی: ژن U7 موشی با استفاده از پرایمرهای اختصاصی از DNA ژنومیک موش با موفقیت تکثیر شد. بررسی محصول PCR نشاندهنده وجود یک باند یکتا با اندازه مورد انتظار بود که بیانگر اختصاصیت بالای پرایمرها و صحت تکثیر ژن هدف است (شکل 1 ب). در مرحله بعد، توالی U7 شامل پروموتر طبیعی، ناحیه کدکننده و توالی انتهایی 3´ به داخل پلاسمید pAAV-trunc بهکمک روش ligase independent cloning (LIC) وارد شد. تعیین توالی پلاسمید نوترکیب نشان داد که توالی U7 بدون هیچگونه حذف، اضافه یا جهش نقطهای در محل صحیح قرار گرفته است. این سازه با نام pAAV-U7 شناسایی شد و بهعنوان مرجع عملکرد طبیعی RNA U7 در ادامه مطالعه مورد استفاده قرار گرفت.
حذف هدفمند دومین اتصال به DNA در پلاسمید pAAV-U7del: برای بررسی نقش دومین اتصال به DNA در عملکرد RNA U7، حذف هدفمند این ناحیه با استفاده از inverse PCR انجام شد. نتایج الکتروفورز محصول PCR نشان داد که پلاسمید حذف شده دارای اندازه کوچکتر نسبت به پلاسمید pAAV-U7 اولیه است که حذف موفق ناحیه هدف را تأیید میکند (شکل 3). نتایج تعیین توالی نیز نشان داد که حذف دومین اتصال به DNA بدون تأثیر بر سایر نواحی ساختاری U7 انجام شده است. پلاسمید حاصل با نام pAAV-U7del بهعنوان بستر مناسب برای ورود توالیهای exon skipping طراحیشده مورد استفاده قرار گرفت.
ساخت و تأیید پلاسمید:pAAV-U7ESex51tail توالی هیبرید سنتتیک شامل ناحیه مکمل ESE اگزون 51 ژن دیستروفین و توالی tailA1 با تمایل اتصال بالا به hnRNPA1 طراحی و سنتز شد. این توالی بهگونهای طراحی شده بود که از طریق همپوشانی انتهایی، امکان اتصال مستقیم به پلاسمید pAAV-U7del را فراهم کند. نتایج کلونینگ با استفاده از روش LIC نشان داد که توالی هیبرید با موفقیت در محل مناسب وارد پلاسمید شده است. بررسی تکثیر PCR و الکتروفورز نشان داد که سازه نهایی کاملاً مطابق طراحی اولیه است (شکل 1ج). پلاسمید حاصل با نام pAAV-U7ESex51tail برای تولید ویروس نوترکیب مورد استفاده قرار گرفت.
تولید و تخلیص ویروس نوترکیبAAVES51: ویروس نوترکیب AAVES51 با کوترانسفکشن پلاسمید pAAV-U7ESex51tail به همراه پلاسمیدهای کمکی در سلولهای HEK293 تولید شد. بررسی مورفولوژی سلولها پس از ترانسفکشن نشاندهنده سلامت نسبی سلولها و عدم بروز سمیت شدید بود. پس از تخلیص ویروس با استفاده از گرادیان سزیم کلرید، ذرات ویروسی با خلوص مناسب برای آلودهسازی سلولی بهدست آمدند.
آلودهسازی سلولهای میوبلاست انسانی با ویروس AAVES51: سلولهای میوبلاست انسانی نامیرا شده با ویروس AAVES51 آلوده شدند. بررسی مورفولوژی سلولی پس از 48 ساعت نشان داد که سلولها زندهمانی بالایی داشته و تغییرات مورفولوژیک قابلتوجهی مشاهده نشد (شکل 2). این یافتهها نشان میدهد که ویروس AAVES51 در شرایط آزمایشگاهی مورد استفاده، اثر سمی قابلتوجهی بر سلولهای میوبلاست انسانی ندارد و برای مطالعات عملکردی مناسب است.
القای حذف اگزون 51 در سطحRNA: نتایج RT-PCR با استفاده از پرایمرهای اختصاصی اطراف اگزون 51 ژن دیستروفین نشان داد که در سلولهای آلودهشده با ویروس AAVES51، باند غالب با طول 210 جفت باز مشاهده شد که بیانگر حذف موفق اگزون 51 است (شکل 3). در مقابل، در نمونههای کنترل غیرآلوده، باند 450 جفت بازی مربوط به حضور اگزون 51 مشاهده شد. این تفاوت آشکار در الگوی باندها نشاندهنده کارایی بالای سیستم U7-ESex51-tail در القای exon skipping هدفمند میباشد.
افزایش بیان پروتئین دیستروفین در سطح پروتئین: نتایج وسترن بلات نشان داد که در سلولهای تیمار شده با ویروس AAVES51، شدت باند پروتئین دیستروفین نسبت به نمونههای کنترل افزایش یافته است (شکل 4). آنالیز دانسیتومتری باندها نیز افزایش قابلتوجه میزان بیان پروتئین دیستروفین را نشان داد. این نتایج بیانگر آن است که حذف اگزون 51 در سطح RNA منجر به بازسازی چهارچوب خوانش ژن دیستروفین و افزایش تولید پروتئین در سطح سلولی شده است.
بررسی حذف اگزون 51 بدنبال آلوده سازی سلول میوبلاست طبیعی با ویروس AAV-ESex51 با استفاده از RT-PCR: آلودهسازی سلولهای میوبلاست طبیعی با ویروس AAV-ESex51 منجر به حذف اگزون 51 از mRNA بالغ دیستروفین شد. بر اساس نتایج بهدست آمده از واکنش RT-PCR بخش قابل توجهی از mRNA استخراج شده از سلول آلوده در مقایسه با سلول کنترل دچار حذف اگزون 51 شده است (شکل3).
***توجه به این نکته ضروری است که بخشی از باند محصول تکثیر توالی واجد اگزون 51 در این واکنش میتواند به علت تکثیر توالیهای hnRNA (نسخه های RNA نابالغ که هنوز تحت فرایند پردازش قرار نگرفتهاند) باشد که نتیجتا راندمان فرایند exon skipping ناشی از آلودهسازی سلول با ویروس AAV-ESex51 احتمالاً بیشتر از آنچه در عکس دیده شده است میباشد.
بررسی حذف اگزون 51 بهدنبال آلودهسازی سلول میوبلاست طبیعی با ویروس AAV-ESex51 با استفاده از وسترن بلات
آلودهسازی سلولهای میوبلاست طبیعی با ویروس AAV-ESex51 منجر به حذف اگزون 51 از mRNA بالغ دیستروفین میشود. این واقعه باعث وقوع جهش تغییر چهارچوب (frameshift) میشود که نهایتا منجر به تخریب RNA و عدم تولید پروتئین دیستروفین میشود (شکل 3).
نتایج حاصل از بررسی وسترن بلات نیز بهطور واضح نشاندهنده کاهش چشمگیر میزان پروتئین دیستروفین در سلولهای آلوده به ویروس میباشد.
شکل 1: نتایج مراحل مختلف تولید AAV تولید کننده توالی U7-ESex51-tail
الف) الکتروفورز ژل آگارز محصول inverse PCR برای حذف کاست بیان ژن نوترکیب از پلاسمید pAAV-MCS و تولید پلاسمید pAAV-trunc نتیجه این واکنش یک باند حدودا 5 کیلوبازی بود که همانطور که در تصویر دیده میشود تکثیر شده بود. ب) تکثیر ژن U7 موشی از DNA ژنومیک موش و بررسی محصول PCR بر روی ژل آگارز. ج) تأیید ورود توالی U7-ESex51-tail به پلاسمید pAAV-U7del با استفاده از PCR الکتروفورز.
شکل 2: بررسی مورفولوژی و آلودهسازی سلولهای میوبلاست انسانی پس از تیمار با ویروس
AAVES51 تصاویر فلورسانس (ستون چپ) نشاندهنده حضور سلولهای آلودهشده با ویروس میباشند، در حالیکه تصاویر میدان روشن (ستون راست) مورفولوژی طبیعی و تراکم مناسب سلولها را پس از تیمار نشان میدهند. عدم مشاهده تغییرات مورفولوژیک بارز بین نمونههای تیمار شده و کنترل بیانگر عدم سمیت قابلتوجه ویروس AAVES51 در شرایط آزمایشگاهی است. (نوار مقیاس: 100 میکرومتر)
شکل 3: نتایج RT-PCR :بررسی حذف اگزون 51 ژن دیستروفین در سلولهای میوبلاست انسانی پس از آلودهسازی با ویروس AAVES51
باند با اندازه تقریبی 210 جفت باز نشاندهنده حذف اگزون 51 در سلولهای تیمار شده است، در حالیکه باند حدودا 450 جفت بازی در نمونه کنترل بیانگر حضور اگزون 51 میباشد. مارکر وزن مولکولی DNA برای تخمین اندازه محصولات PCR استفاده شده است.
شکل 4: نتایج وسترن بلات بررسی بیان پروتئین دیستروفین در سلولهای میوبلاست DMD آلوده شده با ویروس AAVeESex51 در مقابل سلولهای کنترل.
پس از آلودهسازی با ویروس AAVESex51 افزایش شدت باند مربوط به پروتئین دیستروفین (باند بالایی) در نمونه تیمار شده با AAVESex51 نسبت به نمونه کنترل مشاهده میشود. باند پایینی مربوط به ژن خانهدار بهعنوان کنترل داخلی برای نرمالسازی میزان پروتئین بهکار رفته است.
بحث
بیماری دیستروفی عضلانی دوشن (Duchenne Muscular Dystrophy; DMD) یکی از شایعترین و شدیدترین بیماریهای نوروموسکولار وابسته به جنس است که در اثر جهشهای ایجادکننده فریمشیفت در ژن دیستروفین ایجاد میشود (1). از اواسط دهه ۱۹۹۰ میلادی، معرفی مفهوم exon skipping بهعنوان یک راهبرد درمانی مولکولی، افق تازهای را در درمان این بیماری گشود. اساس این رویکرد بر حذف هدفمند یک یا چند اگزون از پیش mRNA استوار است تا چهارچوب خوانش ژن بازسازی شده و یک پروتئین دیستروفین کوتاهتر اما تا حدی عملکردی تولید شود؛ الگویی که از فرم خفیفتر بیماری یعنی دیستروفی عضلانی بیکر(Becker muscular destrophy ) الهام گرفته شده است. ایده استفاده از پدیده پرش اگزونی (Exon Skipping) بهعنوان یک راهبرد درمانی برای دیستروفی عضلانی دوشن (DMD) از اواسط دهه 1990 میلادی مطرح شد و بهسرعت بهعنوان یکی از واقعگرایانهترین رویکردهای درمانی برای این بیماری شناخته شد. منطق زیربنایی این استراتژی بر این اصل استوار است که حذف هدفمند یک یا چند اگزون میتواند چهارچوب خوانش ژن دیستروفین را بازسازی کرده و منجر به تولید یک پروتئین کوتاهشده اما عملکردی، مشابه آنچه در بیماران مبتلا به دیستروفی بکر مشاهده میشود، گردد(9-2). یکی از مهمترین مزایای استراتژی exon skipping در مقایسه با رویکردهای مبتنی بر انتقال DNA نوترکیب، عدم دستکاری مستقیم ژنوم میزبان است. این ویژگی، خطرات بالقوهای نظیر درج تصادفی توالیهای خارجی در ژنوم، فعالسازی انکوژنها یا اختلال در ژنهای حیاتی را بهطور قابلتوجهی کاهش میدهد. به همین دلیل، exon skipping از همان ابتدا بهعنوان رویکردی ایمنتر نسبت به ژندرمانی کلاسیک مطرح شد. در یک دهه گذشته، مطالعات متعددی کارایی و ایمنی این تکنولوژی را در مدلهای سلولی، حیوانی و حتی کارآزماییهای بالینی نشان دادهاند. استفاده از الیگونوکلئوتیدهای آنتیسنس (AONs) مانند eteplirsen که حذف اگزون 51 را القا میکند، به تأیید سازمان FDA نیز رسیده و شواهد بالینی از افزایش نسبی دیستروفین و کند شدن سیر پیشرفت بیماری ارائه داده است (12-11). با این حال، محدودیت اصلی AONهای سنتتیک، نیمهعمر کوتاه، نیاز به تزریقهای مکرر و نفوذ ناکافی به بافت عضله اسکلتی بوده است. برای غلبه بر این محدودیتها، استفاده از ناقلهای ویروسی بهویژه ویروسهای آدنو-مرتبط (AAV) بهعنوان سیستمهای تحویل پایدار مورد توجه قرار گرفت (20).AAVها به دلیل تمایل بافتی بالا به عضله، ایمنی نسبی و توانایی ایجاد بیان پایدارRNA، گزینهای مناسب برای انتقال کاستهای القاکننده exon skipping محسوب میشوند. مطالعات متعددی نشان دادهاند که بیان پایدار RNAهای مبتنی بر U7 snRNA از طریق AAV میتواند منجر به القای طولانیمدت exon skipping در مدلهای mdx شود. در مطالعه حاضر، ما موفق به طراحی و تولید یک وکتور AAV مبتنی بر U7 اصلاحشده شدیم که حذف هدفمند اگزون 51 ژن دیستروفین را القا میکند. نتایج RT-PCR نشان داد که بخش قابلتوجهی از ترانسکریپتهای تولیدشده در سلولهای میوبلاست آلودهشده، فاقد اگزون 51 هستند. این یافته با نتایج مطالعات پیشین که از سیستم U7 برای القای exon skipping استفاده کردهاند، همخوانی دارد و نشاندهنده کارایی مناسب این سیستم در سطح RNA است. از منظر پروتئینی، نتایج وسترن بلات افزایش بیان دیستروفین در سلولهای تیمار شده را نشان داد. این موضوع از نظر بیولوژیکی حائز اهمیت است، زیرا نشان میدهد که حذف اگزون 51 منجر به بازسازی چهارچوب خوانش و ترجمه موفق mRNA اصلاحشده شده است. هرچند میزان دیستروفین تولیدشده به سطح فیزیولوژیک کامل نرسیده، اما شواهد متعددی نشان دادهاند که حتی مقادیر اندک (۵–15%) دیستروفین میتواند اثرات درمانی قابلتوجهی در بیماران DMD داشته باشد. یکی از دستاوردهای مهم این مطالعه، بومیسازی فناوری تولید ناقلهای AAV القاکننده exon skipping در قالب یک پلتفرم انعطافپذیر است. استفاده از تکنولوژی ligase independent cloning (LIC) برای اولین بار در تولید وکتور AAV-U7 در این مطالعه، امکان طراحی سریع، دقیق و کمهزینه وکتورهای متنوع را فراهم کرده است. این موضوع از منظر پزشکی شخصی(personalized medicine) اهمیت ویژهای دارد، زیرا ژن دیستروفین دارای طیف وسیعی از جهشهای حذفی و مضاعفشدگی است و درمان همه بیماران با یک استراتژی واحد امکانپذیر نیست. پلاسمید پایه pAAV-U7del که در این مطالعه طراحی شد، این قابلیت را دارد که بهعنوان یک اسکلت عمومی برای درج سریع توالیهای exon skipping هدفگیرنده اگزونهای مختلف ژن دیستروفین مورد استفاده قرار گیرد. این ویژگی، زمان تولید وکتورهای جدید را به کمتر از یک ماه کاهش داده و هزینه و پیچیدگی فرایند را بهطور چشمگیری کم میکند. علاوه بر این، امکان طراحی وکتورهایی با چندین کاست U7 مستقل وجود دارد که میتواند حذف همزمان چند اگزون را هدف قرار دهد؛ راهبردی که برای بیماران دارای جهشهای متنوع در نواحی hotspot ژن دیستروفین بسیار جذاب است. با وجود مزایای قابلتوجه exon skipping، این رویکرد محدودیتهایی نیز دارد. مهمترین محدودیت آن، عدم اصلاح دائمی ژنوم است؛ بهطوریکه اثر درمانی وابسته به تداوم بیان RNA القاکننده exon skipping باقی میماند. در این زمینه، فناوریهای ویرایش ژن نظیر CRISPR/Cas9 بهعنوان جایگزینهای بالقوه مطرح شدهان CRISPR میتواند با حذف دائمی اگزونهای معیوب یا اصلاح جهشها، درمانی ریشهایتر ارائه دهد. با این حال، سیستم CRISPR نیز با چالشهای مهمی مواجه است؛ از جمله خطر برشهای خارج از هدف (off-target effects)، پاسخهای ایمنی علیه Cas9، و پیچیدگی کنترل دقیق ویرایش در بافتهای مختلف. در مقابل، exon skipping مبتنی بر RNA رویکردی ایمنتر و قابلکنترلتر محسوب میشود که بهویژه در شرایطی که ایمنی اولویت بالاتری دارد، همچنان گزینهای جذاب است. از دیگر محدودیتهای این مطالعه میتوان به استفاده از مدل سلولی بهجای مدل حیوانی، عدم بررسی عملکرد عضلانی و عدم ارزیابی طولانیمدت پایداری بیان دیستروفین اشاره کرد. همچنین، بررسی دقیق میزان exon skipping در سطح کمی (quantitative) و مقایسه با سایر سیستمهای تحویلی میتواند در مطالعات آینده مدنظر قرار گیرد)(16-18).
نتیجهگیری
در مجموع، نتایج این مطالعه نشان میدهد که ترکیب سیستم U7 اصلاحشده با ناقلهای AAV و استفاده از فناوری LIC میتواند یک پلتفرم کارآمد، منعطف و بومی برای توسعه درمانهای exon skipping در بیماری DMD فراهم کند. این رویکرد میتواند بهعنوان پلی میان درمانهای RNA محور و فناوریهای ویرایش ژن، جایگاه ویژهای در آینده درمان دیستروفی عضلانی دوشن داشته باشد.
سپاسگزاری
مطالعه ماحصل طرح تحقیقاتی با شماره 1575 مصوب دانشگاه شهید صدوقی یزد است.
حامی مالی: دانشگاه شهید صدوقی یزد
تعارض در منافع: وجود ندارد.
ملاحظات اخلاقی
پروپوزال این تحقیق توسط دانشگاه علوم پزشکی شهید صدوقی یزد و مشهد تایید شده است.
.مشارکت نویسندگان
دکتر مجید مجرد و دکتر جواد زواررضادر ارائه ایده، و طراحی مطالعه و تجزیه و تحلیل دادهها ، خانم عطیه اصلاحی و حسین تنیپور در انجام کار آزمایشگاهی، جمعآوری دادهها مشارکت داشته و همه نویسندگان در تدوین، ویرایش اولیه و نهایی مقاله و پاسخگویی به سوالات مرتبط با مقاله سهیم هستند.
References:
1. Kesari A, Pirra LN, Bremadesam L, McIntyre O, Gordon E, Dubrovsky AL, et al. Integrated DNA, cDNA, and protein studies in Becker muscular dystrophy show high exception to the reading frame rule. Hum mutat 2008; 29(5): 728-37.
2. Elangkovan N, Dickson G. Gene Therapy for Duchenne Muscular Dystrophy.J Neuromuscul Dis 2021; 8(s2): S303-S316.
3. Tominari T, Aoki Y. Clinical Development of Novel Therapies for Duchenne Muscular Dystrophy—Current and Future. Neurology and Clinical Neuroscience 2022;11(3): 111-18.
4. Gaedigk R, Law DJ, Fitzgerald-Gustafson KM, McNulty SG, Nsumu NN, Modrcin AC, et al. Improvement in Survival and Muscle Function in an Mdx/Utrn(-/-) Double Mutant Mouse Using A Human Retinal Dystrophin Transgene. Neuromuscul Disord 2006;16(3):192-203.
5. Kawai M. Establishment of an Evaluation Method for Muscular Dystrophy and a Patient Registration System for Clinical Trials. Rinsho shinkeigaku 2009; 49(11): 863-6.
6. Tedesco FS. Human Artificial Chromosomes for Duchenne Muscular Dystrophy and Beyond: Challenges and Hopes. Chromosome Res 2015; 23(1): 135-41.
7. Świątkowska‐Flis B, Zdolińska‐Malinowska I, Sługocka D, Boruczkowski D. The Use of Umbilical Cord‐Derived Mesenchymal Stem Cells in Patients with Muscular Dystrophies: Results from Compassionate Use in Real‐Life Settings. Stem Cells Transl Med 2021; 10(10): 1372-83.
8. Koutsoulidou A, Koutalianos D, Georgiou K, Kakouri A, Oulas A, Tomazou M, et al. Serum Mirnas as Biomarkers for the Rare Types of Muscular Dystrophy. Neuromuscular Disorders 2022; 32(4): 332-46.
9. Przymuszała PM, Martyniak A, Kwiatkowska J, Meyer Szary M, Śledzińska K, Wierzba J, et al. Generation of Human Induced Pluripotent Stem Cell Line Derived from Becker Muscular Dystrophy Patient with CRISPR/Cas9-Mediated Correction of DMD Gene Mutation.Stem Cell Res 2024; 76: 103327.
10. Matsuo M, Takeshima Y. Rescue of Dystrophin Mrna of Duchenne Muscular Dystrophy by Inducing Exon Skipping. Acta Myol 2005; 24(2): 110-4.
11. Brolin C, Shiraishi T. Antisense Mediated Exon Skipping Therapy for Duchenne Muscular Dystrophy (DMD). Artif DNA PNA XNA 2011; 2(1): 6-15.
12. Cirak S, Arechavala-Gomeza V, Guglieri M, Feng L, Torelli S, Anthony K, et al. Exon Skipping and Dystrophin Restoration in Patients with Duchenne Muscular Dystrophy after Systemic Phosphorodiamidate Morpholino Oligomer Treatment: An Open-Label, Phase 2, Dose-Escalation Study. Lancet 2011; 378(9791): 595-605.
13. Nakano S, Ozasa S, Yoshioka K, Fujii I, Mitsui K, Nomura K, et al. Exon-Skipping Events in Candidates for Clinical Trials of Morpholino. Pediatr Int 2011; 53(4): 524-9.
14. Hoogaars WM, Mouisel E, Pasternack A, Hulmi JJ, Relizani K, Schuelke M, et al. Combined Effect of AAV-U7-Induced Dystrophin Exon Skipping and Soluble Activin Type IIB Receptor in Mdx Mice. Human Gene Ther 2012; 23(12): 1269-79.
15. Le Hir M, Goyenvalle A, Peccate C, Precigout G, Davies KE, Voit T, et al. AAV Genome Loss From Dystrophic Mouse Muscles During AAV-U7 snRNA-mediated Exon-skipping Therapy. Mol Ther 2013; 21(8): 1551-558.
16. Hart CC, Lee YI, Xie J, Gao G, Lin BL, Hammers DW, et al. Potential Limitations of Microdystrophin Gene Therapy for Duchenne Muscular Dystrophy. JCI Insight 2024; 9(11): e165869.
17. Mendell J, Muntoni F, McDonald C, Mercuri E, Ciafaloni E, et al. AAV Gene Therapy for Duchenne Muscular Dystrophy: The EMBARK Phase 3 Randomized Trial. Nat Med 2025; 31(1): 332-41.
18. Chwalenia K, Feng V, Hemmer N, Friedrichsen H, Vorobieva L, et al. AAV Microdystrophin Gene Replacement Therapy for Duchenne Muscular Dystrophy: Progress and Prospects. Gene Ther 2025; 32(5): 447-61.
19. Chicoine LG, Rodino-Klapac LR, Shao G, Xu R, Bremer WG, Camboni M, et al. Vascular Delivery of Raavrh74.MCK.GALGT2 to the Gastrocnemius Muscle of the Rhesus Macaque Stimulates the Expression of Dystrophin and Laminin Α2 Surrogates. Mol Ther 2014; 22(4): 713-24.
20. Salva MZ, Himeda CL, Tai PW, Nishiuchi E, Gregorevic P, et al. Design of Tissue-Specific Regulatory Cassettes for High-Level Raav-Mediated Expression in Skeletal and Cardiac Muscle. Mol Ther 2007; 15(2): 320-9.
21. Niks EH, Aartsma-Rus A. Exon Skipping: A First in Class Strategy for Duchenne Muscular Dystrophy. Expert Opin Biol Ther 2017; 17(2): 225-36.
22. van Vliet L, de Winter CL, van Deutekom JC, van Ommen GJ, Aartsma-Rus A. Assessment of the Feasibility of Exon 45-55 Multiexon Skipping for Duchenne Muscular Dystrophy. BMC Med Gen 2008; 9: 105.
23. Aslanidis C, de Jong PJ. Ligation-Independent Cloning of PCR Products (LIC-PCR). Nucleic Acids Res 1990; 18(20): 6069-74.
24. Wendrich JR, Liao CY, van den Berg WA, De Rybel B, Weijers D. Ligation-Independent Cloning For Plant Research. Methods Mol Biol 2015; 1284: 421-31.
25. Schmid-Burgk JL, Schmidt T, Hornung V. Ligation-Independent Cloning (LIC) Assembly of TALEN Genes. Methods Mol Biol 2015; 1239: 161-9.
26. Noori-Daloii MR, Mojarrad M, Rashidi-Nezhad A, Kheirollahi M, Shahbazi A, Khaksari M, et al. Use of Sirna in Knocking Down of Dopamine Receptors, A Possible Therapeutic Option in Neuropsychiatric Disorders. Mol biol Rep 2012; 39(2): 2003-10.
27. Nesmith AP, Wagner MA, Pasqualini FS, O'Connor BB, Pincus MJ, August PR, et al. A Human in Vitro Model of Duchenne Muscular Dystrophy Muscle Formation and Contractility. J Cell Biol 2016; 215(1): 47-56.
ارسال پیام به نویسنده مسئول
| بازنشر اطلاعات | |
 |
این مقاله تحت شرایط Creative Commons Attribution-NonCommercial 4.0 International License قابل بازنشر است. |
