

دوره 33، شماره 4 - ( تیر 1404 )
جلد 33 شماره 4 صفحات 8912-8885 |
برگشت به فهرست نسخه ها


![]()
![]()
![]()
Download citation:
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:



BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
Tabrizi F, Khatami M, Heidari M M. Role of Genetic and Epigenetic Factors and Mechanisms Involved in the Occurrence of Congenital Heart Diseases (CHD). JSSU 2025; 33 (4) :8885-8912
URL: http://jssu.ssu.ac.ir/article-1-6377-fa.html
URL: http://jssu.ssu.ac.ir/article-1-6377-fa.html
تبریزی فاطمه، خاتمی مهری، حیدری محمدمهدی. نقش عوامل و مکانیسم های ژنتیکی و اپیژنتیکی دخیل در بروز بیماریهای مادرزادی قلب. مجله علمي پژوهشي دانشگاه علوم پزشكي شهید صدوقی يزد. 1404; 33 (4) :8885-8912



متن کامل [PDF 1456 kb]
(312 دریافت)
| چکیده (HTML) (973 مشاهده)
References:
1- Bernier P-L, Stefanescu A, Samoukovic G, Tchervenkov CI. The Challenge of Congenital Heart Disease Worldwide: Epidemiologic and Demographic Facts. Seminars in Thoracic and Cardiovascular Surgery: Pediatric Cardiac Surgery Annual 2010; 13(1): 26-34.
2- Zhao L, Chen L, Yang T, Wang T, Zhang S, Chen L, et al. Birth Prevalence of Congenital Heart Disease in China, 1980–2019: A Systematic Review and Meta-Analysis of 617 Studies. Eur J Epidemiol 2020; 35(7): 631-42.
3- Shieh JTC, Bittles AH, Hudgins L. Consanguinity and the Risk of Congenital Heart Disease. American J Med Genetics Part A 2012; 158A (5): 1236-41.
4- Zaidi S, Brueckner M. Genetics and Genomics of Congenital Heart Disease. Circ Res 2017; 120(6): 923-40.
5- Wang X, Li P, Chen S, Xi L, Guo Y, Guo A, et al. Influence of Genes and the Environment in Familial Congenital Heart Defects. Mol Med Rep 2014; 9(2): 695-700.
6- Sun R, Liu M, Lu L, Zheng Y, Zhang P. Congenital Heart Disease: Causes, Diagnosis, Symptoms, and Treatments. Cell Biochemistry and Biophysics 2015; 72(3): 857-60.
7- Zhu H, Kartiko S, Finnell R. Importance of Gene–Environment Interactions in the Etiology of Selected Birth Defects. Clinical Genetics 2009; 75(5): 409-23.
8- Bragança J, Pinto R, Silva B, Marques N, Leitão HS, Fernandes MT. Charting the Path: Navigating Embryonic Development to Potentially Safeguard against Congenital Heart Defects. J Personalized Med 2023; 13(8): 1263.
9- Blue GM, Kirk EP, Sholler GF, Harvey RP, Winlaw DS. Congenital Heart Disease: Current Knowledge About Causes and Inheritance. Med J Australia 2012; 197 (3): 155-59.
10- Lage K, Greenway SC, Rosenfeld JA, Wakimoto H, Gorham JM, Segrè AV, et al. Genetic and Environmental Risk Factors in Congenital Heart Disease Functionally Converge in Protein Networks Driving Heart Development. Proceedings of the National Academy of Sciences 2012; 109(35): 14035-40.
11- Roberts AE, Lacro RV. Genetics of Congenital Heart Disease. In Nadas' Pediatric Cardiology. 3ed. Elsevier, 2025; 55-63.
12- LaHaye S, Corsmeier D, Basu M, Bowman JL, Fitzgerald-Butt S, Zender G, et al. Utilization of Whole Exome Sequencing to Identify Causative Mutations in Familial Congenital Heart Disease. Circulation: Cardiovascular Genetics 2016; 9(4): 320-9.
13- Zahavich L, Bowdin S, Mital S. Use of Clinical Exome Sequencing in Isolated Congenital Heart Disease. Circulation: Cardiovascular Genetics 2017; 10(3): e001581.
14- Page DJ, Miossec MJ, Williams SG, Monaghan RM, Fotiou E, Cordell HJ, et al. Whole Exome Sequencing Reveals the Major Genetic Contributors to Nonsyndromic Tetralogy of Fallot. Circulation Research 2019; 124(4): 553-63.
15- Jin SC, Homsy J, Zaidi S, Lu Q, Morton S, DePalma SR, et al. Contribution of Rare Inherited and De Novo Variants In 2,871 Congenital Heart Disease Probands. Nature Genetics 2017; 49(11): 1593-601.
16- Peterlin A, Bertok S, Writzl K, Lovrečić L, Maver A, Peterlin B, et al. The Genetic Architecture of Congenital Heart Disease in Neonatal Intensive Care Unit Patients, The Experience of University Medical Centre, Ljubljana. Life 2024; 14(9): 1118.
17- Majumdar U, Yasuhara J, Garg V. In Vivo and in Vitro Genetic Models of Congenital Heart Disease. Cold Spring Harbor Perspectives in Biology 2021; 13(4): a036764.
18- Lin H, McBride KL, Garg V, Zhao MT. Decoding Genetics of Congenital Heart Disease Using Patient-Derived Induced Pluripotent Stem Cells (iPSCs). Front Cell Dev Biol 2021; 9: 630069.
19- Guo H, Liu L, Nishiga M, Cong L, Wu JC. Deciphering Pathogenicity of Variants of Uncertain Significance with CRISPR-Edited Ipscs. Trends in Genetics 2021; 37(12): 1109-23.
20- Khairy P, Ionescu-Ittu R, Mackie AS, Abrahamowicz M, Pilote L, Marelli AJ. Changing Mortality in Congenital Heart Disease. J American College of Cardiology 2010; 56(14): 1149-57.
21- Zimmerman MS, Smith AGC, Sable CA, Echko MM, Wilner LB, Olsen HE, et al. Global, Regional, and National Burden of Congenital Heart Disease: A Systematic Analysis for the Global Burden of Disease Study 2017. Lancet Child Adolescent Health 2020; 4(3): 185-200.
22- Shabana NA, Shahid SU, Irfan U. Genetic Contribution to Congenital Heart Disease (CHD). Pediatr Cardiol 2020; 41(1): 12-23.
23- Suluba E, Shuwei L, Xia Q, Mwanga A. Congenital Heart Diseases: Genetics, Non-Inherited Risk Factors, and Signaling Pathways. Egyptian J Med Human Genetics 2020; 21(1): 11.
24- Evans PR. Cardiac Anomalies in Mongolism. British Heart Journal 1950; 12(3): 258-62.
25- Nawaz K, Alifah N, Hussain T, Hameed H, Ali H, Hamayun S, et al. From Genes to Therapy: A Comprehensive Exploration of Congenital Heart Disease Through the Lens of Genetics and Emerging Technologies. Current Problems in Cardiology 2024; 49(9): 102726.
26- Moyer AJ, Gardiner K, Reeves RH. All Creatures Great and Small: New Approaches for Understanding Down Syndrome Genetics. Trends in Genetics 2021; 37(5): 444-459.
27- Irving CA, Chaudhari MP. Cardiovascular Abnormalities in Down Syndrome: Spectrum, Management and Survival Over 22 Years. Arch Dis Child 2012; 97(4): 326-30.
28- Pfitzer C, Helm PC, Rosenthal L-M, Berger F, Bauer UMM, Schmitt KRL. Dynamics in Prevalence of Down Syndrome in Children with Congenital Heart Disease. Europ J Pediatrics 2018; 177(1): 107-15.
29- Narayan P, Richter F, Morton S. Genetics and Etiology of Congenital Heart Disease. Curr Top Dev Biol 2024; 156: 297-331.
30- Obler D, Juraszek AL, Smoot LB, Natowicz MR. Double Outlet Right Ventricle: Aetiologies and Associations. J Med Genet 2008; 45(8): 481-97.
31- Qiao F, Wang Y, Zhang C, Zhou R, Wu Y, Wang C, et al. Comprehensive Evaluation of Genetic Variants Using Chromosomal Microarray Analysis and Exome Sequencing in Fetuses with Congenital Heart Defect. Ultrasound Obstet Gynecol 2021; 58 (3): 377-87.
32- Costain G, Silversides CK, Bassett AS. The Importance of Copy Number Variation in Congenital Heart Disease. NPJ Genom Med 2016; 1(1): 16031.
33- Zhao Y, Diacou A, Johnston HR, Musfee FI, McDonald-McGinn DM, McGinn D, et al. Complete Sequence of the 22q11.2 Allele in 1,053 Subjects with 22q11.2 Deletion Syndrome Reveals Modifiers of Conotruncal Heart Defects. Am J Hum Genet 2020; 106(1): 26-40.
34- Škorić-Milosavljević D, Lahrouchi N, Bosada FM, Dombrowsky G, Williams SG, Lesurf R, et al. Rare Variants in KDR, Encoding VEGF Receptor 2, Are Associated with Tetralogy of Fallot. Genetics in Medicine 2021; 23(10): 1952-1960.
35- Duran I, Tenney J, Warren CM, Sarukhanov A, Csukasi F, Skalansky M, et al. NRP1 Haploinsufficiency Predisposes to the Development of Tetralogy of Fallot. American Journal of Medical Genetics Part A 2018; 176(3): 649-56.
36- Glessner JT, Bick AG, Ito K, Homsy JG, Rodriguez-Murillo L, Fromer M, et al. Increased Frequency of De Novo Copy Number Variants in Congenital Heart Disease by Integrative Analysis of Single Nucleotide Polymorphism Array and Exome Sequence Data. Circulation Research 2014; 115(10): 884-96.
37- Soemedi R, Wilson Ian J, Bentham J, Darlay R, Töpf A, Zelenika D, et al. Contribution of Global Rare Copy-Number Variants to the Risk of Sporadic Congenital Heart Disease. Am J Human Genet 2012; 91(3): 489-501.
38- Teekakirikul P, Zhu W, Gabriel GC, Young CB, Williams K, Martin LJ, et al. Common Deletion Variants Causing Protocadherin; Deficiency Contribute to the Complex Genetics of BAV and Left-Sided Congenital Heart Disease. Human Genetics and Genomics Advances 2021; 2(3): 100037.
39- Griffin EL, Nees SN, Morton SU, Wynn J, Patel N, Jobanputra V, et al. Evidence-Based Assessment of Congenital Heart Disease Genes to Enable Returning Results in a Genomic Study. Circ Genom Precis Med 2023; 16 (2): e003791.
40- Landis BJ, Helvaty LR, Geddes GC, Lin JHI, Yatsenko SA, Lo CW, et al. A Multicenter Analysis of Abnormal Chromosomal Microarray Findings in Congenital Heart Disease. J Am Heart Assoc 2023; 12 (18): e029340.
41- Boskovski MT, Homsy J, Nathan M, Sleeper LA, Morton S, Manheimer KB, et al. De Novo Damaging Variants, Clinical Phenotypes, and Post-Operative Outcomes in Congenital Heart Disease. Circ Genom Precis Med 2020; 13(4): e002836.
42- Falsaperla R, Giacchi V, Aguglia MG, Mailo J, Longo MG, Natacci F, et al. Monogenic Syndromes with Congenital Heart Diseases in Newborns (Diagnostic Clues for Neonatologists): A Critical Analysis with Systematic Literature Review. J Pediatr Genet 2021; 10(3): 173-93.
43- Khatami M, Ghazinader D, Ahmadi F, Heidari MM, Hadadzadeh M, Namnabat M. Novel Missense Mutation in NKX2.6 Gene (C.389 G > C, Arg130Pro) As A Potentially Pathogenic Variant in Pediatric Patients with Congenital Heart Disease. Gene Reports 2023; 33: 101819.
44- Hsieh A, Morton SU, Willcox JAL, Gorham JM, Tai AC, Qi H, et al. EM-Mosaic Detects Mosaic Point Mutations that Contribute to Congenital Heart Disease. Genome Med 2020; 12(1): 42.
45- Boyle L, Wamelink MMC, Salomons GS, Roos B, Pop A, Dauber A, et al. Mutations in TKT Are the Cause of a Syndrome Including Short Stature, Developmental Delay, And Congenital Heart Defects. Am J Hum Genet. 2016; 98(6): 1235-42.
46- Pinna V, Daniele P, Calcagni G, Mariniello L, Criscione R, Giardina C, et al. Prevalence, Type, And Molecular Spectrum of NF1 Mutations in Patients with Neurofibromatosis Type 1 and Congenital Heart Disease. Genes 2019; 10(9): 675.
47- Teekakirikul P, Zhu W, Xu X, Young CB, Tan T, Smith AM, et al. Genetic Resiliency Associated with Dominant Lethal TPM1 Mutation Causing Atrial Septal Defect with High Heritability. Cell Rep Med 2022; 3(2): 100501.
48- Helle E, Córdova-Palomera A, Ojala T, Saha P, Potiny P, Gustafsson S, et al. Loss of Function, Missense, And Intronic Variants in NOTCH1 Confer Different Risks for Left Ventricular Outflow Tract Obstructive Heart Defects in Two European Cohorts. Genetic Epidemiol 2019; 43(2): 215-26.
49- Stephen J, Maddirevula S, Nampoothiri S, Burke JD, Herzog M, Shukla A, et al. Bi-allelic TMEM94 Truncating Variants Are Associated with Neurodevelopmental Delay, Congenital Heart Defects, and Distinct Facial Dysmorphism. The American Journal of Human Genetics 2018; 103(6): 948-67.
50- Heidari MM, Khatami M, Kamalipour A, Kalantari M, Movahed M, Emmamy MH, et al. Mitochondrial Mutations in Protein Coding Genes of Respiratory Chain Including Complexes IV, V, and Mt-Trna Genes Are Associated Risk Factors for Congenital Heart Disease. Excli j 2022; 21: 1306-30.
51- Khatami M, Heidari MM, Karimian N, Hadadzadeh M. Mitochondrial Mutations in tRNAGlu and Cytochrome b Genes Associated with Iranian Congenial Heart Disease. Int Cardiovasc Res J 2016; 10(4): e9815.
52- Canac R, Cimarosti B, Girardeau A, Forest V, Olchesqui P, Poschmann J, et al. Deciphering Transcriptional Networks during Human Cardiac Development. Cells 2022; 11(23): 3915.
53- Dianatpour S, Khatami M, Heidari MM, Hadadzadeh M. Novel Point Mutations of CITED2 Gene are Associated with Non-Familial Congenital Heart Disease (CHD) in Sporadic Pediatric Patients. Appl Biochem Biotechnol 2020; 190(3): 896-906.
54- Khatami M, Heidari MM, Kazeminasab F, Zare Bidaki R. Identification of A Novel Non-Sense Mutation in TBX5 Gene in Pediatric Patients with Congenital Heart Defects. J Cardiovasc Thorac Res 2018; 10(1): 41-5.
55- Tabrizi F, Khatami M, Heidari MM, Bragança J, Tatari H, Namnabat M, et al. Novel and Deleterious Nucleotide Variations in the HAND1 Gene Probably Affect Mirna Target Sites and Protein Function in Pediatric Patients with Congenital Heart Disease. Mol Biol Reports 2024; 51(1): 468.
56- Gonzalez-Teran B, Pittman M, Felix F, Thomas R, Richmond-Buccola D, Hüttenhain R, et al. Transcription Factor Protein Interactomes Reveal Genetic Determinants in Heart Disease. Cell 2022; 185(5): 794-814.e730.
57- Wu Y, Jin X, Zhang Y, Zheng J, Yang R. Genetic and Epigenetic Mechanisms in the Development of Congenital Heart Diseases. World J Pediatr Surg 2021; 4(2): e000196.
58- MacGrogan D, Münch J, de la Pompa JL. Notch and Interacting Signalling Pathways in Cardiac Development, Disease, And Regeneration. Nat Rev Cardiol 2018; 15(11): 685-704.
59- Reuter MS, Jobling R, Chaturvedi RR, Manshaei R, Costain G, Heung T, et al. Haploinsufficiency of Vascular Endothelial Growth Factor Related Signaling Genes Is Associated with Tetralogy of Fallot. Genet Med 2019; 21(4): 1001-7.
60- Williams K, Carson J, Lo C. Genetics of Congenital Heart Disease. Biomolecules 2019; 9(12): 879.
61- Hanna A, Frangogiannis NG. The Role of the TGF-β Superfamily in Myocardial Infarction. Front Cardiovasc Med 2019; 6.
62- Stefanovic S, Zaffran S. Mechanisms of Retinoic Acid Signaling during Cardiogenesis. Mech Dev 2017; 143: 9-19.
63- Nakajima Y. Retinoic Acid Signaling in Heart Development. Genesis 2019; 57 (7-8): e23300.
64- Zaffran S, Robrini NE, Bertrand N. Retinoids and Cardiac Development. J Develop Biology 2014; 2(1): 50-71.
65- Wang G, Wang B, Yang P. Epigenetics in Congenital Heart Disease. J Am Heart Assoc 2022; 11(7): e025163.
66- Coppola A, Romito A, Borel C, Gehrig C, Gagnebin M, Falconnet E, et al. Cardiomyogenesis Is Controlled by the Mir-99a/Let-7c Cluster and Epigenetic Modifications. Stem Cell Res 2014; 12(2): 323-37.
67- Linglart L, Bonnet D. Epigenetics and Congenital Heart Diseases. J Cardiovasc Dev Dis 2022; 9(6): 185.
68- Lim TB, Foo SYR, Chen CK. The Role of Epigenetics in Congenital Heart Disease. Genes (Basel) 2021; 12(3): 390.
69- Wang G, Wang B, Yang P. Epigenetics in Congenital Heart Disease. J Am Heart Assoc 2022; 11(7): e025163.
70- Wu Y, Jin X, Zhang Y, Zheng J, Yang R. Genetic and Epigenetic Mechanisms in the Development of Congenital Heart Diseases. World J Pediatr Surg 2021; 4(2): e000196.
71- Gilsbach R, Preissl S, Grüning BA, Schnick T, Burger L, Benes V, et al. Dynamic DNA Methylation Orchestrates Cardiomyocyte Development, Maturation and Disease. Nat Commun 2014; 5: 5288.
72- Asim A, Agarwal S, Panigrahi I, Saiyed N, Bakshi S. MTHFR Promoter Hypermethylation May Lead to Congenital Heart Defects in Down Syndrome. Intractable & Rare Diseases Research 2017; 6(4): 295-8.
73- Ho L, Crabtree GR. Chromatin Remodeling during Development. Nature 2010; 463(7280): 474-84.
74- Hang CT, Yang J, Han P, Cheng H-L, Shang C, Ashley E, et al. Chromatin Regulation by Brg1 Underlies Heart Muscle Development and Disease. Nature 2010; 466 (7302): 62-7.
75- Chang S, McKinsey TA, Zhang CL, Richardson JA, Hill JA, Olson EN. Histone Deacetylases 5 and 9 Govern Responsiveness of the Heart to a Subset of Stress Signals and Play Redundant Roles in Heart Development. Mol Cell Biol 2004; 24(19): 8467-76.
76- Park CY, Pierce SA, von Drehle M, Ivey KN, Morgan JA, Blau HM, et al. Sknac, A Smyd1-Interacting Transcription Factor, Is Involved in Cardiac Development and Skeletal Muscle Growth and Regeneration. Proc Natl Acad Sci USA 2010; 107(48): 20750-5.
77- Consortium EP. An Integrated Encyclopedia of DNA Elements in the Human Genome. Nature 2012; 489(7414): 57-74.
78- Chung I-M, Rajakumar G. Genetics of Congenital Heart Defects: The NKX2-5 Gene, a Key Player. Genes 2016; 7(2): 6.
79- Fotiou E, Williams S, Martin-Geary A, Robertson DL, Tenin G, Hentges KE, et al. Integration of Large-Scale Genomic Data Sources with Evolutionary History Reveals Novel Genetic Loci for Congenital Heart Disease. Circ Genom Precis Med 2019; 12(10): 442-51.
80- Nees SN, Chung WK. Genetic Basis of Human Congenital Heart Disease. Cold Spring Harbor Perspectives in Biology 2020; 12(9): a036749.
81- Touma M. Genome Regulation by Long Noncoding Rnas in Neonatal Heart Maturation and Congenital Heart Defects. J Clin and Mol Med 2020; 3(1): 1-6.
82- Dickel DE, Barozzi I, Zhu Y, Fukuda-Yuzawa Y, Osterwalder M, Mannion BJ, et al. Genome-Wide Compendium and Functional Assessment of in Vivo Heart Enhancers. Nat Commun 2016; 7: 12923.
83- Smemo S, Campos LC, Moskowitz IP, Krieger JE, Pereira AC, Nobrega MA. Regulatory Variation in a TBX5 Enhancer Leads to Isolated Congenital Heart Disease. Human Mol Genetics 2012; 21(14): 3255-63.
84- Richter F, Morton SU, Kim SW, Kitaygorodsky A, Wasson LK, Chen KM, et al. Genomic Analyses Implicate Noncoding De Novo Variants in Congenital Heart Disease. Nature Genetics 2020; 52(8): 769-77.
85- Lahm H, Jia M, Dreßen M, Wirth F, Puluca N, Gilsbach R, et al. Congenital Heart Disease Risk Loci Identified by Genome-Wide Association Study in European Patients. J Clin Invest 2021; 131(2): e141837.
86- Cordell HJ, Bentham J, Topf A, Zelenika D, Heath S, Mamasoula C, et al. Genome-Wide Association Study of Multiple Congenital Heart Disease Phenotypes Identifies a Susceptibility Locus for Atrial Septal Defect at Chromosome 4p16. Nat Genet 2013; 45(7): 822-4.
87- Agopian A, Goldmuntz E, Hakonarson H, Sewda A, Taylor D, Mitchell LE. Genome-Wide Association Studies and Meta-Analyses for Congenital Heart Defects. Circ Cardiovasc Genet 2017; 10(3): e001449
88- Yu M, Aguirre M, Jia M, Gjoni K, Cordova-Palomera A, Munger C, et al. Oligogenic Architecture of Rare Noncoding Variants Distinguishes 4 Congenital Heart Disease Phenotypes. Circ Genom Precis Med 2023;16(3): 258-66.
89- Khera AV, Chaffin M, Aragam KG, Haas ME, Roselli C, Choi SH, et al. Genome-Wide Polygenic Scores for Common Diseases Identify Individuals with Risk Equivalent to Monogenic Mutations. Nature Genetics 2018; 50(9): 1219-24.
90- Ameres SL, Zamore PD. Diversifying Microrna Sequence and Function. Nat Rev Mol Cell Biol 2013; 14(8): 475-88.
91- Fischer SEJ. RNA Interference and MicroRNA-Mediated Silencing. Curr Protoc Mol Biol 2015; 112: 26.21.21-26.21.25.
92- Diener C, Keller A, Meese E. The Mirna–Target Interactions: An Underestimated Intricacy. Nucleic Acids Res 2023; 52(4): 1544-57.
93- Sempere LF, Freemantle S, Pitha-Rowe I, Moss E, Dmitrovsky E, Ambros V. Expression Profiling of Mammalian Micrornas Uncovers a Subset of Brain-Expressed Micrornas with Possible Roles in Murine and Human Neuronal Differentiation. Genome Biol 2004; 5(3): R13.
94- Lagos-Quintana M, Rauhut R, Yalcin A, Meyer J, Lendeckel W, Tuschl T. Identification of Tissue-Specific Micrornas from Mouse. Curr Biol 2002; 12(9): 735-9.
95- Callis TE, Pandya K, Seok HY, Tang RH, Tatsuguchi M, Huang ZP, et al. Microrna-208a Is a Regulator of Cardiac Hypertrophy and Conduction in Mice. J Clin Invest 2009; 119(9): 2772-86.
96- Liu N, Williams AH, Kim Y, McAnally J, Bezprozvannaya S, Sutherland LB, et al. An Intragenic MEF2-Dependent Enhancer Directs Muscle-Specific Expression of Micrornas 1 and 133. Proc Natl Acad Sci USA 2007; 104(52): 20844-9.
97- Zhao Y, Samal E, Srivastava D. Serum Response Factor Regulates a Muscle-Specific Microrna that Targets Hand2 during Cardiogenesis. Nature 2005; 436(7048): 214-20.
98- McCarthy JJ. Microrna-206: The Skeletal Muscle-Specific Myomir. Biochim Biophys Acta 2008; 1779(11): 682-91.
99- Dueñas A, Expósito A, Aranega A, Franco D. The Role of Non-Coding Rna in Congenital Heart Diseases. J Cardiovasc Dev Dis 2019; 6(2): 15.
100- Khatami M, Ghorbani S, Adriani MR, Bahaloo S, Naeini MA, Heidari MM, et al. Novel Point Mutations in 3′-Untranslated Region of GATA4 Gene Are Associated with Sporadic Non-Syndromic Atrial and Ventricular Septal Defects. Curr Med Sci 2022; 42(1): 129-43.
101- Clowes C, Boylan M, Ridge L, Barnes E, Wright J, Hentges K. The Functional Diversity of Essential Genes Required for Mammalian Cardiac Development. Genesis 2014; 52(8): 713-37.
102- Kalayinia S, Arjmand F, Maleki M, Malakootian M, Singh CP. Micrornas: Roles in Cardiovascular Development and Disease. Cardiovasc Pathol 2021; 50: 107296.
103- Anderson KM, Anderson DM. Lncrnas at the Heart of Development and Disease. Mamm Genome 2022; 33(2): 354-65.
104- Alexanian M, Ounzain S. Long Noncoding RNAs in Cardiac Development. Cold Spring Harb Perspect Biol 2020; 12(11): a037374.
105- Martens L, Rühle F, Witten A, Meder B, Katus HA, Arbustini E, et al. A Genetic Variant Alters the Secondary Structure of the Lncrna H19 and is Associated with Dilated Cardiomyopathy. RNA Biol 2021; 18(sup1): 409-15.
106- Sweta S, Dudnakova T, Sudheer S, Baker AH, Bhushan R. Importance of Long Non-Coding Rnas in the Development and Disease of Skeletal Muscle and Cardiovascular Lineages. Front Cell Devel Biol 2019; 7: 228.
متن کامل: (883 مشاهده)
مقدمه
بیماری مادرزادی قلب یا Congenital Heart Disease (CHD) بهعنوان یک ناهنجاری آناتومیکی شدید در بافت قلب یا عروق بزرگ مرتبط با آن تعریف میشود که از لحاظ اثر بر عملکرد طبیعی بدن اهمیت دارد. CHD شایعترین ناهنجاری مادرزادی در سراسر جهان است که حدود 1 درصد از تولدهای زنده را تحت تأثیر قرار میدهد. این بیماری که علت اصلی مرگومیر ناشی از نقصهای مادرزادی است، منجر به سقط حدود 10 درصد از جنینها در دوران بارداری میشود. بهطور جهانی، تقریباً حدود ۲/۱ میلیون نفر در سال، با نقصهای گسترده قلبی متولد میشوند (1,2). علل زمینهای بروز CHD نسبتاً کمتر شناخته شده است و ترکیبی از عوامل ژنتیکی، اپیژنتیکی و محیطی در ایجاد آن نقش دارند. شواهد اپیدمیولوژیک بر تأثیر غالب عوامل ژنتیکی تأکید دارند، از جمله افزایش خطر ابتلا در افرادی با سابقه خانوادگی، تطابق بیشتر در دوقلوهای همسان نسبت به غیرهمسان، و شیوع بالاتر در جمعیتهایی با ازدواجهای فامیلی (6-3). با اینحال، درصد قابلتوجهی از موارد بیماری، در خانوادههای بدون سابقه قبلی از CHD رخ میدهد که نشاندهنده وقوع جهشهای جدید (de novo)، مانند ناهنجاریهای کروموزومی (8%-12%)، واریانتهای تعداد نسخه ژنی یا Copy Number Variant (CNV) (3%-25%) و جهشهای نقطهای (3%-5%) است (4). در مقابل، عوامل غیرژنتیکی شناختهشده، شامل تراتوژنهای محیطی، مواجهات مادر با عوامل خطر در دوران بارداری و عوامل عفونی که 8%-12% کل موارد بیماران را تشکیل میدهند (7). تخمین زده میشود در حدود 400 ژن در پاتوژنز CHD نقش داشته باشند. جهش در ژنهای کدکننده پروتئینهای مرتبط با بازسازی (رمدلینگ) کروماتین، سیگنالینگ سلولی و فاکتورهای رونویسی میتوانند فرآیند تمایز سلولهای قلبی را مختل کرده و به اختلالات ساختاری و عملکردی قلب منجر شوند (8). با اینحال، علل ژنتیکی CHD، در تقریبا 80% موارد ناشناخته است. این بیماری به دلیل تنوع ژنتیکی و هتروژنسیتی پیچیده (حالتی که جهشهای ژنی مختلفی باعث بیماری میشوند)، غالبا الگوهای وراثتی مندلی را دنبال نمیکند. این هتروژنسیتی منجر به درصد نفوذ و بیان متغیر ژنهای دخیل میشود (11-9). اگرچه ژنهای کاندید متعددی برای بروز CHD سندرمی شناسایی شدهاند، بررسی حالات CHD غیرسندرمی، به دلیل هتروژن بودن آن چالشبرانگیزتر است. امروزه پیشرفتهای چشمگیر در تکنولوژیهای توالییابی DNA، شناسایی واریانتهای نادر در ژنهای جدید مرتبط با CHD غیرسندرمی را نیز ممکن ساخته است (15-12). تکنیکهایی مانند توالییابی نسل جدید یا Next-generation sequencing (NGS)، توالییابی RNA تکسلولی (Single Cell RNA sequencing)، توالییابی microRNA (miRNA sequencing) و آنالیز سلولهای بنیادی پرتوان القایی انسانی (hiPSCs) Human-Induced Pluripotent Stem Cell در شناسایی علل و سازوکارهای بیماری CHD بسیار مؤثر بودهاند (16-18). علاوه براین، مطالعه و بررسی بیماریزا بودن واریانتهای جدید و با اهمیت نامشخص یا Variants of Uncertain Significance (VUS) با استفاده از سلولهای iPSC ویرایششده توسط CRISPR امکانپذیر شده است (19). این تکنیکها با ارزیابی نقایص عملکردی واریانتهای خاص، کیفیت زندگی نوزادان مبتلا به CHD را بهبود بخشیدهاند، بهطوری که بیش از 90% آنها به سنین بزرگسالی میرسند (20). مطالعات مختلفی برای بررسی علتشناسی بیماریهای مادرزادی قلب انجام شده است. با اینحال، شناسایی عوامل مولکولی و مکانیسمهای مرتبط با این بیماری، همچنان موضوع بحث و تحقیقات متخصصین است. درک علل ژنتیکی منجر شونده به CHD میتواند بینش عمیقی در مورد زیربنای مولکولی این بیماری فراهم کند و در تعریف تخمین ریسک بیماری و بهبود روشهای پیشگیری از آن مؤثر باشد. همچنین، چنین دانشی میتواند برنامهریزی برای درمانهای نوین را تسهیل کند و اساس زیستشناسی تکوین قلب را روشنتر نماید و قادر است به پیشبینی نتایج بالینی، ارزیابی خطر در خانوادهها و غربالگری افراد در معرض عوامل خطر کمک کند. علاوه براین، مطالعهی ارتباطات ژنوتیپ-فنوتیپ در این بیماری، میتواند اطلاعات ارزشمندی درباره پیشآگهی (prognosis) بیماری CHD ارائه دهد. این مقاله مروری به جدیدترین یافتهها درباره زیربنای ژنتیکی این بیماری میپردازد.
روش بررسی
برای دستیابی به نتایج این پژوهش مروری، نخستین گام یک جستجوی جامع در پایگاههای دادهای معتبر علمی، از جمله PubMed، Scopus و ScienceDirect بود. در این مرحله، با استفاده از کلیدواژههای تخصصی مرتبط با موضوع مانند «congenital heart defect»،«congenital heart disease»، «genetic basis»،«epigenetic modifications»، «molecular mechanisms»، و دیگر عبارات جستجوهایی طراحی و اجرا شدند تا متون علمی مرتبط شناسایی گردد. این جستجوها بهگونهای انجام شدند که هم مطالعات قدیمیتر و هم پژوهشهای منتشرشده در سالهای اخیر مورد بررسی قرار گیرند و بدین وسیله، تصویر جامع و بهروزی از مبنای ژنتیکی و اپیژنتیکی CHD ارائه شود. پس از شناسایی مقالات، گزینش آنها بر اساس معیارهای پیشتعیینشده صورت گرفت. از جمله این معیارها میتوان به جدیدبودن یافتهها (انتشار در سالهای اخیر)، وجود اعتبار علمی (داشتن روند داوری علمی و انتشار در نشریات معتبر) و پوشش موضوع اشاره کرد. مقالاتی که معیارهای مورد اشاره را نداشتند، از انتخاب حذف شدند. در مرحله بعد، اطلاعات کلیدی از ۸۶ مقاله منتخب استخراج گردید. این اطلاعات شامل یافتههای مهم در زمینههای مختلف از جمله واریانتهای ژنتیکی، تغییرات متنوع و متعدد اپیژنتیکی و مکانیسمهای مولکولی مربوط به CHD بود. فرآیند استخراج دادهها به دقت و بهصورت دستی انجام شد تا از صحت و دقت اطلاعات اطمینان حاصل شود. سپس، دادههای استخراجشده دستهبندی و تحلیل شدند. این تحلیل شامل بررسی نقاط مشترک و تفاوتهای میان مطالعات و تبیین ارتباط بین یافتههای بهدستآمده بود. هدف از دستهبندی فاکتورهای ژنتیکی و اپیژنتیکی مورد بررسی، ارائه یک دیدگاه جامع و انتقادی نسبت به همه فاکتورهای ژنتیکی و اپیژنتیکی دخیل در ایجاد CHD بود که به انعکاس وضعیت دانش موجود در این حوزه، و همچنین خلأهای پژوهشی بپردازد و به مطرحشدن پیشنهادهایی در مورد موضوع جهتگیریهای تحقیقات آتی کمک نماید. در نهایت، دادههای طبقهبندی شده در قالب یک دیدگاه یکپارچه و علمی ارائه شدند.
آمارهای جمعیتی و دستهبندی بیماریهای مادرزادی قلبی: بیماریهای مادرزادی قلب یا Congenital Heart Disease (CHD) یک طیف گسترده از ناهنجاریهای ساختاری قلب هستند که در دوران رشد رویان ایجاد میشوند و از عوامل مهم در بروز بیماری و مرگ و میر در کودکان هستند. با وجود پیشرفتهای عظیم در تشخیص و مدیریت بیماریهای قلبی مادرزادی در چند دهه گذشته، بیشتر اطلاعات موجود درباره مدیریت بیماریهای قلبی مادرزادی از مناطقی با مقدار بالای شاخص اجتماعی- جمعیتشناختی یا Socio-demographic Index (SDI) بهدست آمده است. با وجود محدودیت جمعآوری داده، از کشورهای با SDI پائین، «آنالیز سیستماتیک بیماریهای مادرزادی قلب- Global Burden of Disease (GBD) 2017» جامعترین ارزیابی جهانی از آمار بیماریهای مادرزادی قلب تا به امروز را ارائه میدهد. این آنالیزها که حاصل همکاری پژوهشی چندین مرکز تحقیقاتی هستند، توسط مؤسسه Institute for Health Metrics and Evaluation (IHME) تجمیع و در سال ۲۰۲۰ منتشر شده است. این دادهها توسط متخصصان، پزشکان، آموزشدهندگان، محققان و سیاستگذاران در سراسر جهان برای کنترل بیماری CHD، تدوین سیاستهای بهداشتی مؤثر و گرفتن تصمیمات کلیدی در رابطه با بهبود زندگی بیماران استفاده میشود (21). گزارش GBD به ارزیابی بروز و مرگومیر بیماری CHD، آسیبهای ناشی از بیماری و عوامل خطر بیماریها در سطح جهانی میپردازد. طبق دادههای اپیدمیولوژیک ارائه شده، تخمین زده شده است که در سال ۲۰۱۷ تقریباً 12 میلیون نفر در جهان مبتلا به بیماریهای مادرزادی قلب بودهاند که نشاندهنده افزایش ۷/۱۸ درصدی نسبت به سال 1990 میباشد. تعداد مرگومیرهای مرتبط با بیماری در سال 2017 حدود ۲۶۱ هزار نفر برآورد شده است که کاهش ۵/۳۴ درصدی نسبت به تعداد تخمین زده شده در سال 1990 دارد. بیماریهای مادرزادی قلبی، بهعنوان یکی از علل اصلی مرگومیر نوزادان شناخته میشوند که در سطح جهانی رتبه ششم و در مناطق با SDI بالا، رتبه دوم را دارا هستند. با تغییر علل اصلی مرگومیر از بیماریهای واگیردار به بیماریهای غیرواگیردار، انتظار میرود اهمیت بیماریهای قلبی مادرزادی، به عنوان یکی از عوامل اصلی مرگومیر نوزادان در سطح جهانی در سالهای آینده بیشتر مورد توجه قرار گیرد (21). شکل ۱ جدیدترین آمار میزان مرگومیر ناشی از بیماریهای مادرزادی قلبی در هر ۱۰۰۰۰۰ نفر طبق گزارش GBD سال ۲۰۲۰ در سراسر جهان به تفکیک هر کشور را نمایش میدهد. سیستم کدگذاری بینالمللی برای بیماریهای مادرزادی قلب یا International Paediatric and Congenital Cardiac Code (IPCCC) که شامل۳۶۷ عنوان بیماری استاندارد است، یک چارچوب طبقهبندی شده برای این ناهنجاریها ارائه میدهد. این سیستم توسط انجمن بینالمللی نامگذاری و سازمان بهداشت جهانی، در ویرایش یازدهم از «دستهبندی جهانی طبقهبندی بیماریها» یا International Classification of Diseases 11th Revision (ICD-11) معرفی شده است. طبق این طبقهبندی استاندارد، بیماریهای مادرزادی قلب بهطور کلی به چهار دسته اصلی تقسیم میشوند. نقایص هیپوپلازی (Hypoplasia defects) یکی از این دستههاست که در آن تکامل یک طرف از قلب ناقص میماند و توانایی پمپاژ خون در آن سمت مختل میشود. بسته به سمت درگیر، این ناهنجاری به دو شکل اصلی، شامل سندرم قلب چپ هیپوپلاستیک یا Hypoplastic Left Heart Syndrome (HLHS) و سندرم قلب راست هیپوپلاستیک یا Hypoplastic Right Heart Syndrome (HRHS) دیده میشود. دسته دوم، نقایص انسدادی یا Obstructive Heart Defects (OHDs)، شامل ناهنجاریهایی است که مسیر جریان خون در دریچهها، سیاهرگها یا سرخرگها را تنگ یا مسدود میکنند. از جمله نمونههای این دسته، میتوان به کوارکتاسیون آئورت یا Coarctation of Aorta (CoA) و تنگی آئورت یا Aortic Stenosis (AS) اشاره کرد. دسته سوم، نقایص دیوارهای (Septal Defect)، شایعترین نوع بیماریهای مادرزادی قلب هستند. این ناهنجاریها به دلیل نقص یا سوراخ در دیوارههای قلب ایجاد میشوند که منجر به اختلال در جریان طبیعی خون بین حفرههای قلب میشوند. نقص دیواره بین بطنی یا Ventricular septal defect (VSD) و نقص دیواره بین دهلیزی یا Atrial Septal Defect (ASD) از جمله نمونههای رایج این دسته هستند. در نهایت، نقایص سیانوتیک یا Cyanotic Heart Disease به ناهنجاریهایی گفته میشود که باعث کاهش اکسیژنرسانی به بافتها شده و معمولاً با کبودی (سیانوز) همراه هستند. این دسته شامل مواردی مانند اتصال نابجای همه وریدهای ریوی یا Total Anomalous Pulmonary Venous Connection (TAPVC)، تترالوژی فالو یا Tetralogy of Fallot (ToF)، جابجایی سرخرگهای بزرگ یا Transposition of the Great Arteries (TGA) و تنه مشترک سرخرگی پایا یاPersistent Truncus Arteriosus (PTA) است. جدول ۱ شماری از شایعترین انواع بیماریهای مادرزادی قلب در نوزادان را نمایش میدهد. طبقهبندی دقیق این بیماریها کمک میکند تا پزشکان ناهنجاریهای قلبی مادرزادی را بهتر تشخیص داده و درمان مناسب را برنامهریزی کنند. علاوه بر این، این دستهبندی استاندارد به جمعآوری دادههای دقیق برای تحقیقات و پیشگیری کمک شایانی میکند (22).
عوامل ژنتیکی موثر در بروز CHD: پیشرفت در تکنیکهای مولکولی، امکان مطالعه نقصهای تکوینی قلب را فراهم کرده و دانش محققین را از مورفولوژی بیماری و عوامل ژنتیکی دخیل در آن افزایش داده است. شواهد بسیاری نشان میدهند که عوامل ژنتیکی، نقش کلیدی در پاتوژنز CHD ایفا میکنند. جهشهای نقطهای در ژنهای فاکتورهای رونویسی قلب، پلیمورفیسمهای تکنوکلئوتیدی یا Single-Nucleotide Polymorphism (SNP)، آنیوپلوئیدیهای کروموزومی (Chromosomal aneuploidies)، واریانتهای تعداد نسخه کروموزومی یا Copy Number Variation (CNV) و جهش در ژنهای گیرندهها و لیگاندهای مربوط به مسیرهای پیامرسانی مورفوژنز قلب، همگی از عوامل ژنتیکی مرتبط با بروز CHD هستند. جهشها و تغییرات ژنتیکی در این عوامل رونویسی، با بسیاری از CHDهای غیرسندرمی مرتبط است. مطالعات عملکردی بر روی این ژنها در آزمایشات حیوانی نشاندهنده نتایج قابلاعتماد و تکرارپذیری بوده است که مدل توارث تکژنی برای پاتوژنز CHD را پیشنهاد میدهند. با اینحال، مدل توارث تکژنی دو سؤال کلیدی و مهم را مطرح میکند: اول آنکه، چرا انواع مختلفی از CHD با یک نوع جهش ژنی دیده میشوند؟ دوم آنکه، چرا جهشهای متفاوت تکژنی باعث ایجاد فنوتیپهای مشابه CHD میشوند؟ این پرسشها نشان میدهند که احتمالاً عوامل متعدد و مدل توارث چندعاملی در علتشناسی CHD دخیل هستند (23). یکی از جنبههای جالب توجه CHD این است که با وجود کاهش پتانسیل تولیدمثلی در بیماران مبتلا، شیوع این بیماری در جمعیتها ثابت باقی مانده است. این امر احتمال رخداد جهشهای نوظهور را مطرح میکند. با اینحال، انتظار میرود که روند انتخاب طبیعی، باعث کاهش شیوع CHD شود، زیرا این بیماریها با میزان مرگومیر بالا و کاهش توان تولیدمثل مرتبط هستند. همچنین، هنوز مشخص نیست که CHD دارای الگوی توارث مشخصی باشد و اینکه آیا بهصورت غالب اتوزومی و یا مغلوب اتوزومی به ارث میرسد. جهشهای غالب اتوزومی معمولاً با نفوذ بالایی همراه هستند و انتظار میرود درصد زیادی از بستگان درجهیک، CHD را به ارث ببرند. با اینحال، مشاهدات نشان میدهند که این فرضیه همیشه صحیح نیست. بهنظر میرسد الگوی مغلوب اتوزومی برای توضیح اساس ژنتیکی CHD مناسبتر باشد. با توجه به هتروژن بودن این بیماری، مطالعات همراهی سراسر ژنوم یا Genome-wide association studies (GWAS) ابزار ارزشمندی برای شناسایی عوامل ژنتیکی متعددی است که به بروز CHD کمک میکنند (23).
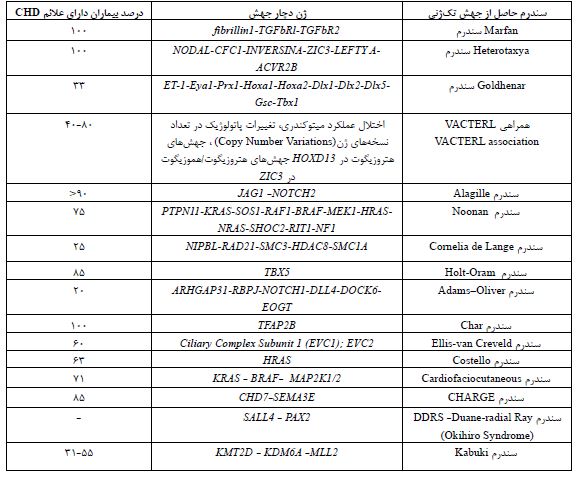
آنیوپلوئیدیهای کروموزومی (Chromosomal Aneuploidies): آنیوپلوئیدیهای کروموزومی از اولین علل ژنتیکی شناختهشدهCHD هستند (24). آنیوپلوئیدیها، تغییراتی در تعداد کروموزومها هستند که معمولاً به صورت de novo (نوظهور- یعنی از قبل در والدین وجود ندارند) و از طریق ناهنجاری در تقسیمات میوزی یا میتوزی ایجاد میشوند. اصلیترین تریزومیهای اتوزومی موجود در جمعیت انسانی، یعنی تریزومی کروموزومهای ۱۳، ۱۸ و ۲۱، با CHD مرتبط هستند، همانطور که سندرم ترنر (مونوزومی کروموزوم X- Turner syndrome) نیز چنین است. در جدول ۲، درصد بیماران دارای فنوتیپ CHD و همچنین انواع CHD دیده شده در چهار سندرم آنیوپلوئیدی مورد اشاره آورده شده است. سندرمهای ناشی از انواع آنیوپلوئیدیها، نفوذپذیری کاملی دارند، اما شدت بیان CHD در آنها متغیر است. به عنوان مثال، تنها در ۴۰ الی ۵۰ درصد افراد با تریزومی ۲۱ (کامل یا جزئی)، CHD از نوع نقص دیواره دهلیزی-بطنی (Atrioventricular Septal Defect) و در بخشی از مبتلایان به سندرم ترنر، تنگی آئورت (Coarctation of the Aorta) مشاهده میشود. با وجود اینکه علت ژنتیکی CHD مرتبط با آنیوپلوئیدیها شناخته شده است، مکانیسمهای مولکولی که باعث اختلال در تکوین قلب میشوند و منجر به نفوذپذیری متغیر CHD میشوند، هنوز بهطور کامل توضیح داده نشدهاند. تحقیقات جدید برای درک این مکانیسمها، بر دستورزی ژنها و مهندسی ژنتیک متمرکز هستند. آنیوپلوئیدیها به ندرت در کشت سلولی مورد مطالعه قرار میگیرند، زیرا سلول واجد آنها در کشت سلولی زنده نمیماند و این موضوع باعث میشود بیشتر پژوهشهای مرتبط با CHD در آنیوپلوئیدیها به مدلهای جانوری و مدلهای انسانی (مانند ارگانوئیدها) متکی باشند (26،27). سازمان ملی ثبت بیماریهای قلبی مادرزادی آلمان (kompetenznetz-ahf.de) که بزرگترین دفتر ثبت CHD در اروپا است، با استفاده از کد بینالمللی IPCCC (International Pediatric and Congenital Cardiac Code) برای دستهبندی انواع CHD و همچنین آنالیز بیماران سندرم داون (Down syndrome) نشان داده است که شیوع انواع CHDها از قبیل تترالوژی فالو، نقص دیوارهی دهلیزی-بطنی، نقص دیوارهی بین دهلیزی و نقص دیوارهی بین بطنی، در افراد مبتلا به تریزومی 21 همواره بالا بوده است (28،29). علاوه بر مطالعات ثبت (Register Study)، روش دیگر برای توصیف ارتباط ژنوتیپ-فنوتیپ در آنیوپلوئیدیها؛ بررسی یک فنوتیپ خاص و سپس توصیف ارتباطات آن با دلایل و علل ژنتیکی است. برای مثال، یک گروه تحقیقاتی، از طریق بررسی گسترده متون علمی نشان دادند که بیماری خروجی دوگانه از بطن راست یا Double outlet right ventricle (DORV) بهطور معمول با تریزومیهای 13 و 18 مرتبط است (30). به دلیل آنکه آنیوپلوئیدیها بهطور معمول در دوران بارداری مورد غربالگری قرار میگیرند، زنان باردار بالای 35 سال، به دلیل ارتباط شیوع بالاتر آنیوپلوئیدیها با افزایش سن مادر، همگی تحت اکوی قلبی جنینی نیز قرار میگیرند. این بررسی میتواند از طریق آمنیوسنتز، تست غیرتهاجمی پیش از تولد یا Non-invasive prenatal testing (NIPT)، کاریوتایپ، آنالیز میکروآرایه (Microarray analysis)، و توالییابی نسل جدید یا Next-Generation Sequencing (NGS) انجام شود (31).
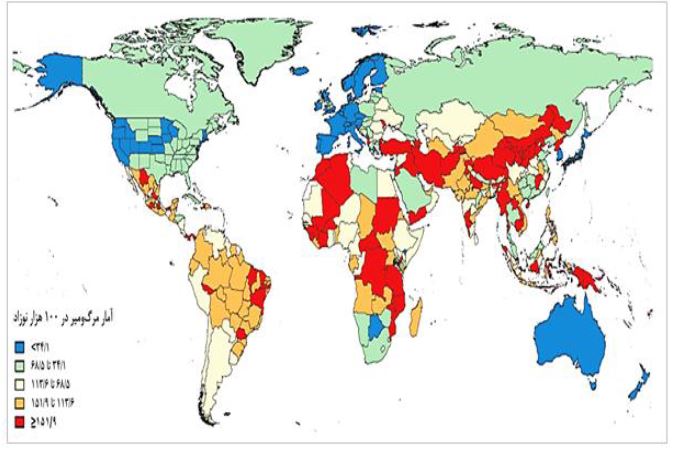
شکل ۱. جدیدترین آمار میزان مرگ و میر نوزادان ناشی از بیماریهای مادرزادی قلبی طبق گزارش GBD سال ۲۰۲۰.
کشور ایران، از مناطق با میزان SDI متوسط تا بالا، جزو مناطق با بالاترین امار مرگو میر ناشی ازین بیماری (رنگ قرمز) نمایش داده شده است (21).
جدول۱: توصیف و دستهبندی شایعترین بیماریهای مادرزادی قلب در کودکان
جدول۲: سندرمهای آنیوپلوئیدی که فنوتیپ CHD در مبتلایان آنها دیده میشود (25)
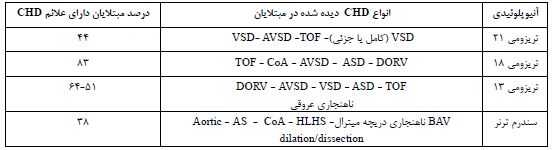
اختصارات؛ VSD: نقص دیواره بین بطنی، TOF: تترالوژی فالو، AVSD: نقص دیواره دهلیزی-بطنی، CoA: تنگی آئورت، ASD: نقص دیواره بین دهلیزی، DORV: خروجی دوگانه از بطن راست، BAV: دریچه آئورت دو لتی، HLHS: سندروم هیپوپلازی قلب چپ، AS: تنگی دریچه آئورت، TGA: جابجایی عروق بزرگ، PS: تنگی دریچه ریوی.
تغییرات تعداد نسخه CNV: واریانتهای تعداد نسخه یا Copy Number Variation (CNV) از جمله ناهنجاریهای ساختاری کروموزومها هستند و حداقل 10-12 درصد از موارد بیماری قلبی مادرزادی را شامل میشوند (32). سندرمهای ناشی از CNVهای با علائمCHD میتوانند به صورت de novo یا ارثی ایجاد شوند و در جمعیت عمومی بسیار نادر هستند. تاکنون، تغییرات ساختاری مرتبط با CHD در مطالعات خانوادگی و گروهی (cohort) مورد بررسی قرار گرفتهاند. سندرمهای CNV ارثی، ممکن است در والدین ناقل بدون علامت باشند و در جمعیت عمومی مشاهده شوند، بنابراین احتمالاً عوامل ژنتیکی، اپیژنتیکی و محیطی وجود دارند که منجر به نفوذپذیری متغیر بیماری میشوند (29). یکی از سندرمهای CNVمهم، حذف ناحیهی کروموزومی 22q11.2 است که باعث سندرم ولوکاردیوفاشیال (Velocardiofacial syndrome) یا سندرم دیجورج (DiGeorge syndrome) میشود. حذف کروموزومی در ناحیه 22q11.2، دومین علت شایع CHDسندرمی پس از تریزومی 21 است و به تنهایی حدود 4 درصد از موارد CHD را شامل میشود. با این حال، همه افراد دارای سندرم حذف در ناحیه 22q11.2، علائم CHD را ندارند (معمولاً تنها 60-70 درصد موارد دارای علایم بیماری هستند) که نشاندهندهی نیاز به تحقیقات بیشتر در مورد اثر تعدیلکنندههای ژنتیکی و غیرژنتیکی (genetic and non-genetic modifiers) بر نفوذ بیماری CHD است (33). مطالعات مورد-شاهدی CHD خانوادگی میتوانند CNVهای ارثی مرتبط با خطر CHD را شناسایی کنند. برای مثال، در یک خانواده با تکرار موارد بیماری تترالوژی فالو بین اعضا آن خانواده، حذف 1/8 مگابازی در موقعیت کروموزومی 10p11 شناسایی شد. این موقعیت کروموزومی دربرگیرنده ژن مهمی است که گیرنده همکار فاکتور رشد اندوتلیال عروقی یا Vascular endothelial growth factor (VEGF) co-receptor به نام نوروپیلین ۱ یا Neuropilin-1 (NRP1) را کد میکند. در سلولهای آمنیوسیت، یکی از اعضای این خانوادهی مبتلا، سطح کاهشیافته بیان ژن NRP1 نیز مشاهده شد (34،35). علاوه براین، با مقایسهی مناطق حذف کروموزومی در افراد مبتلا به سندرم حذف 10q26، ژن WDR11 به عنوان یک ژن کاندید جدید برای سه فنوتیپ بیماری CHD، بیماری کلوبوما (Coloboma، نقص مادرزادی ساختار چشم که به دلیل بسته نشدن کامل شکاف جنینی در دوران رشد ایجاد میشود) و تأخیر رشدی جنین شناسایی شد. کشف واریانتهای تک نوکلئوتیدی در این ژن که همپوشانی فنوتیپی با سندرمهای حذفی در همان ژن را دارند، همانند نقش تائید شده ژن MEIS2، میتواند نقش آن ژن در بروز CHD را اثبات کند. پروتئین کدشده توسط این ژن به نام Meis Homeobox 2 متعلق به خانواده homeobox میباشد که در تنظیم بیان ژنهای مهمی در طی تکوین رویانی نقش دارد. چنین مطالعاتی، مناطق ژنومی کاندید را شناسایی میکنند که ممکن است ردههای سلولی تکوینی متعددی را تحت تاثیر قرار دهند (34). همچنین مشخص شده است که حذفهای هموزیگوت در ژن PCDHA که کدکننده پروتئین پروتوکادهرین آلفا (Protocadherin α) میباشد با بیماری انسداد مجرای خروجی بطن چپ یا Left ventricular outflow tract obstruction (LVOTO) مرتبط است، به طوری که این تغییرات ساختاری کروموزومی، در نیمی از گروه مورد مطالعه مبتلا به تنگی آئورت ایزوله و در ۱۶ درصد از افراد گروه کنترل مشاهده شد. در بررسی آرایههای پلیمورفیسم تکنوکلئوتیدی (single nucleotide polymorphism arrays) در 2539 فرد مبتلا به CHD و 1538 فرد بدون CHD، در مبتلایان افزایش 1/8 برابری در حذفهای نادر نواحی کدکننده مشاهده شد. همچنین در ناحیه کروموزومی 15q11.2 حذفهایی در ژنهای مرتبط با مسیر پیامرسانی Wnt گزارش شد (38-36). بنابراین، تغییرات ساختاری که بهطور حتم با CHD مرتبط هستند، شامل حذف یا تکثیر در ناحیه کروموزومی 1q21.1، حذف 1p36، حذف یا تکثیر 7q11.23، حذف یا تکثیر 8p23، حذف 10q، حذف 11qter حذف 15q11.2، حذف 15q25.2، حذف یا تکثیر 16p11.2 و حذف یا تکثیر22q11 هستند (41-39). جدول ۳ سندرمهای مرتبط با تغییرات تعداد نسخه، درصد بیماران دارای علائم CHD و ژن یا ناحیه کروموزومی دچار تغییر را نمایش میدهد. واریانتهای تک ژنی یا Single Gene Variants (SGVs): این گروه از واریانتها در ژنهای مرتبط با سندرمهای تکژنی از جمله؛ فاکتورهای رونویسی، عوامل پیامرسان داخل سلولی، مولکولهای چسبندگی سلولی و پروتئینهای ساختاری قلب رخ میدهند (43-25). بیماریهای قلبی مادرزادی که ناشی از نقصهای ژنی تکگانه با الگوهای وراثتی مندلی هستند و اغلب بهصورت سندرمی ظاهر میشوند، سهم کوچکی از انواع بیماری CHD در اثر عوامل ژنتیکی را تشکیل میدهند. واریانتهای نوظهور، حدود ۸ درصد از کل موارد CHD را شامل میشوند، در حالیکه تغییرات تکنوکلئوتیدی اتوزومال مغلوب یا single-nucleotide variants (SNVs) و جهشهای درجی- حذفی (indels) تنها حدود ۲ درصد از این موارد را در بر میگیرند (15). اگرچه بسیاری از علل مونوژنیک CHD هنوز شناسایی نشدهاند، مطالعه ژنهای شناختهشده مرتبط با CHD و محصولات پروتئینی آنها، بینشهای ارزشمندی را درباره تکوین و زیستشناسی قلب فراهم کرده است. نکتهی مهم این است که بیشتر توالییابیهای ژنوم انسان در نمونههای خون محیطی بیماران انجام میشود که این روش ممکن است تغییرات موزائیک یا سوماتیک موجود در بافتهای قلبی را شناسایی نکند (44). مطالعات ژنتیکی انسانی با استفاده از تحلیلهای کشف ژنهای جدید و مرتبط، درک ما را از واریانتهای نادر در نواحی کدکنندهی پروتئین مرتبط با CHD بهبود بخشیدهاند. جدول ۴ سندرمهای مرتبط با CHD که در اثر جهشهای تکژنی نقطهای بهوجود آمدهاند، ژن دچار جهش در آن سندرم و درصد بیماران دارای فنوتیپ CHD را نمایش میدهد. این تحلیلها شامل مطالعات همراهی سراسر ژنوم یا بررسی فهرستهای ویژهای از ژنهای کاندید هستند (29). به عنوان مثال، واریانتهای نادر متعدد در ژنهای NOTCH1 و FLT4 در افراد مبتلا به تترالوژی فالو یافت شده است (14). افزایش استعداد ابتلا به CHD با واریانتهای نادر در ژنهای مرتبط با سندرمهای ژنتیکی و فنوتیپهای دیگر غیر از فنوتیپ نقایص قلبی مانند ژن POLR1A (در اختلالات جمجه-صورت)، ژن KDM2B (در اختلالات تکوین سیستم عصبی) و ژن PPP2R1A (در اختلالات تکوین سیستم عصبی) شناسایی شده است. همچنین، با توجه به همزمانی قابل توجه بروز CHD و اختلالات تکوین سیستم عصبی، واریانت ژن TKT، کدکنندهی ترنس کتولاز (Transketolase) در افراد خانوادهای با ویژگیهای تأخیر رشدی، کوتاهقدی و CHD شناسایی شد (45). وجود واریانت در گروههای مبتلا به نوروفیبروماتوز نوع ۱ یا سندرم Ellis-Van Creveld نیز ارتباطات خاص ژنوتیپ-فنوتیپ با ناهمگنی فنوتیپی یک سندرم را تائید مینماید (46). برخی از مطالعات بر روی ژنهای خاص شناساییشده در بیماریهای قلبی مادرزادی خانوادگی تمرکز کرده و از تفاوتهای فنوتیپی اعضا برای بررسی اساس چندژنی این بیماری استفاده میکنند. بهعنوان مثال، یک حذف نادر کوچک در ژن TPM1، که کدکننده زنجیرهی آلفا-تروپومیوزین (α-tropomyosin) است، در یک خانواده با نقص دیواره دهلیزی که در پنج نسل متوالی مشاهده شده بود، از طریق آنالیز پیوستگی (Linkage Analysis) در سطح ژنوم شناسایی شد. قبل انجام این آنالیز، با استفاده از توالییابی کل ژنوم واریانتهای خاص پیدا شده بود. کاهش بیان Tpm1 در طول تکوین، از طریق تکنیک مورفولینو (Morpholino antisense oligonucleotide) و کریسپر یا Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) در مدل Xenopus tropicalis، منجر به ادم قلبی و نقص دیوارهی دهلیزی شد. حذف ژنی Tpm1کدکننده زنجیرهی آلفا-تروپومیوزین (α-tropomyosin) با روشCRISPR-Cas9 در موشها، با خمیدن غیرطبیعی لولهی قلبی اولیه و مرگ زودهنگام رویان همراه بود، که ضرورت وجود آلفا-تروپومیوزین در تکوین قلب مهرهداران را تائید میکند (47). ژن NOTCH1 نیز که گیرنده پروتئین Notch (Notch Receptor 1) را کد میکند، مثال دیگری است که واریانتهای آن طی تکنیک توالییابی اگزوم به روش paired-end در مبتلایان به سندرم قلب چپ هیپوتروفیک که سایر ناهنجاریهای مادرزادی را نداشتند، شناسایی شد (48). واریانتهای نادر مغلوب، مانند جهشهای دوگانه در NKX2-6 (کد کننده پروتئین هومئوباکس Nkx-2.6 یا Homeobox protein Nkx-2.6) و TMEM94 (کد کننده پروتئین تراغشایی ۹۴ یا Transmembrane protein 94) نیز بهعنوان علل ایجاد CHD شناسایی شدهاند (49). نقص در پروتئین TMEM94 به عنوان یک علت شناختهشده برای اختلالات تکوینی سیستم عصبی شناخته میشود (49)، اما در اعضای چند خانواده دارای واریانت این ژن، فنوتیپ بیماری مادرزادی قلب هم وجود داشت. روش توالییابی اگزوم نیز دو واریانت متمایز در این ژن را در هر افراد مبتلا به CHD شناسایی کرد که توارث دوآللی داشتند. همچنین، توالییابی RNA از رده سلولی لنفوبلاستوئید (Lymphoblastoid cell lines) از اعضای یک خانوادهی مبتلا، تأثیر واریانتهای محل پیرایش (splice site variants) بر بروز CHD را تأیید کرد (49). علاوه بر مطالعات ژنومی روی گروههای انسانی، سلولهای بنیادی پرتوان القاشده یا Induced Pluripotent Stem Cells (iPSC) میتوانند بهعنوان مدل in vitro برای بررسی تکوین قلب استفاده شوند. بهعنوان مثال، واریانت بدمعنی p.R443P در پروتئین MYH6 از زیرواحدهای پروتئین میوزین (Myosin)، که به عنوان واریانت بیماریزا و مرتبط با سندرم قلب چپ هیپوتروفیک شناسایی شده بود، در یک تحقیق با استفاده از CRISPR-Cas9 و نوترکیبی همولوگ در iPSCهای انسانی ایجاد شد تا تمایز این سلولها به کاردیومیوسیت یا سلول ماهیچه قلبی (cardiomyocyte) تحت اثر این واریانت مورد مطالعه قرار بگیرد. این سلولهای بنیادی پرتوان القایی به کاردیومیوسیتها تبدیل شدند و با استفاده از CHIR99021 و Activin-A (دو ترکیب شیمیایی مورد استفاده برای حفظ ویژگی بنیادی سلول) و در محیط RPMI/B27 بدون انسولین کشت داده شدند. پس از اصلاح واریانت p.R443P در پروتئین MYH6، بهبود تمایز کاردیومیوسیتها و ساختار سارکومر در رده سلولی فرد پروباند مشاهده شد. این مطالعه بر اهمیت سلولهای بنیادی پرتوان القایی مشتق از بیماران و ویرایش ژنومی آنها در ارزیابی عملکرد بیماریزایی واریانتهای پرخطر بیماری قلبی مادرزادی تأکید میکند (29). جهشهایی در ژنوم میتوکندری در مبتلایان به بیماریهای مادرزادی قلبی نیز شناسایی شدهاند که ممکن است در بیماریزایی این اختلالات قلبی دخیل باشند (50،51).
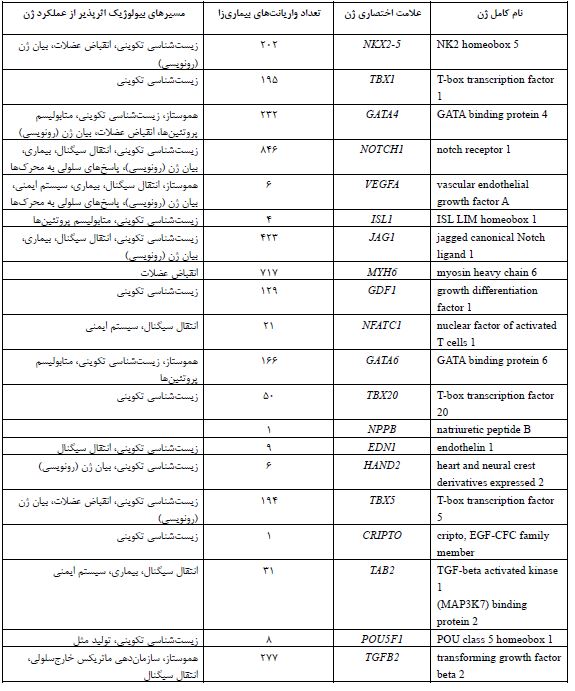
فاکتورهای رونویسی اختصاصی قلب (Cardiac Transcription Factors): تشکیل قلب یک فرآیند پیچیده و نیازمند تعامل بین انواع سلولهای متمایز و وابسته، از طریق مسیرهای پیامرسانی و مسیرهای رونویسی در مکان و زمان خاص است که به تمایز و تعیین سرنوشت آنسلولها منجر میشود (شکل ۲-الف و ۲-ب). اختلال در این فرآیند تکوینی میتواند باعث بروز بیماریهای مادرزادی قلب شود. برنامه تنظیم بیان ژنهای اختصاصی تکوین قلب، توسط فاکتورهای رونویسی که بیان ژنهای کدکننده فاکتورهای رونویسی دیگر را تنظیم کرده و شبکههای خاصی مانند GATA4، NKX2-5 و TBX5 را تشکیل میدهند، هدایت میشود. جهشهای بیماریزا در هر یک از فاکتورهای رونویسی مهم در تکوین رویانی قلب، موجب بر همخوردن این برنامهی بسیار دقیق و پیچیده و ایجاد انواع نقصهای ساختاری قلب خواهد شد (43، 55-52). جدول ۵، بیست مورد از فاکتورهای رونویسی با بالاترین امتیاز «ارتباط ژن-بیماری یا Gene-Disease Interaction (gdi)» را به همراه تعداد واریانتهای بیماریزای آنها نمایش داده است. اختلال در دو فاکتور رونویسی اختصاصی و ضروری تکوین قلب، به نامهای GATA4 و TBX5 از اولین علل ژنتیکی شناختهشدهی CHD خانوادگی میباشند. واریانتهای بیماریزای هتروزیگوت در TBX5 باعث نقص در جدایی حفرهها و تشکیل دیواره و سایر اشکال CHD در سندرم هولت-اورام (Holt-Oram) میشوند. همچنین، واریانتهای هتروزیگوت در ژن GATA4 باعث نقصهای دیواره دهلیزی- بطنی، تنگی شریان ریوی، و ناهنجاری مجراهای خروجی قلب میشوند. اختلال در برهمکنش فیزیکی بین این دو پروتئین یا برهمکنش با سایر کوفاکتورهای اختصاصی در اثر رخداد واریانتهای بدمعنی در آنها، باعث ناهنجاریهای شدید قلبی میشود (56).
جدول۳ : سندرمهای ناشی از ریزحذف و تغییرات تعداد کپی یا CNV همراه با فنوتیپ CHD (9،22،49).
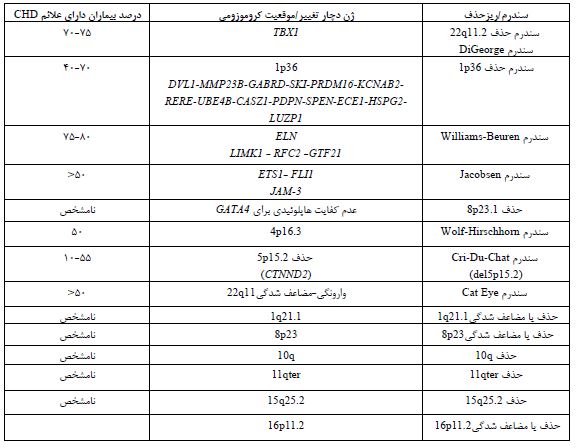
جدول۴: سندرمهای مرتبط با CHD که در اثر جهشهای تک ژنی نقطهای بهوجود آمدهاند (9،22،42).
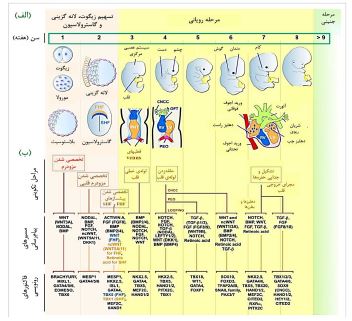
شکل ۲: تکوین قلب انسان (الف) نمایش شماتیک مراحل تکوین انسان و نقاط عطف کلیدی آن. سلولهای ناحیه اولیه قلب FHF(آبی) و ناحیه ثانویه قلب SHF(نارنجی) سلولهای تاج عصبی قلبی CNCCs(سبز) و اندام پرو اپیکاردیال PEO (بنفش) و بخشهای مشتق از آنها به تصویر کشیده شدهاند. در هفته دوم، مزودرم قلبی به پیشسازهای FHF و SHF تبدیل میشود که ساختار هلال قلب را تشکیل میدهند. FHF لوله اولیه قلب (PHT) را تولید میکند که به ترتیب به بطن چپ (LV) و قسمتهایی از دهلیزهای راست و چپ تبدیل میشود. سلولهای SHF در رشد بطن راست (RV)، مجرای خروجی (OFT)، دهلیزها و میوکارد ورودی نقش دارند. سلولهای PEO در تشکیل اپیکارد و رگهای کرونری مشارکت دارند. سلولهای CNCCs از لوله عصبی پشتی به OFT قلب مهاجرت میکنند و در تشکیل دیواره، جدایی شریانهای آئورت و ریوی و تشکیل دریچهها و توزیع عصب پاراسمپاتیک نقش دارند. (ب) مروری بر مسیرهای اصلی پیامرسانی و مهمترین عوامل رونویسی که هر مرحله ذکر شده از تکوین قلب را تنظیم میکنند. مسیر غیرکلاسیک WNT با ncWNT نشان داده شده است (8).
جدول۵: فاکتورهای رونویسی قلبی مهم که به ترتیب بیشترین امتیاز ارتباط ژن- بیماری مادرزادی قلبی (gdi) را دارا هستند. تعداد بسیاری از دیگر فاکتورهای رونویسی دخیل در CHD در این جدول نمایش داده نشدهاند.
مسیرهای پیامرسانی داخل سلولی: شبکهای بسیار دقیق و هوشمند، شامل مسیرهای پیامرسانی Nodal، Transforming Growth Factor-β (TGFβ)، Bone Morphogenetic Protein (BMP)، WNT و رتینوئیک اسید (Retinoic acid)، فرآیند فعالسازی بیان ژنهای مرتبط با فاکتورهای رونویسی قلبی را هدایت میکنند. این فاکتورها، از جمله NKX2-5، GATA4/5/6، و TBX1/5/20، برای تبدیل سلولهای پیشساز نواحی اولیه و ثانویه قلب به سلولهای تخصصی مرتبط با حفرههای مختلف قلب ضروری میباشند (57). مسیر پیامرسانی Notch در فرآیند تکوین دریچههای قلب و حفظ هموستازی آنها نقش حیاتی ایفا میکنند (58). فاکتور پروتئینی رشد اندوتلیال عروقی یا Vascular endothelial growth factor A (VEGF-A) به عنوان یک میتوژن کلیدی، در تشکیل دیواره دریچههای دهلیزی- بطنی، مانند دریچههای تریکاسپید (tricuspid valve) و میترال (mitral valve) نقش دارد. تغییر در سطح بیان این پروتئین در مراحل تکوین قلب، چه افزایشی باشد و چه کاهشی، با نقصهای مادرزادی قلبی مانند نقص دیواره دهلیزی- بطنی مرتبط است (59). همچنین، در مسیر پیامرسانی Hedgehog بیان ژن Sonic hedgehog (SHH) برای تشکیل دیوارهی بین بطنی قلب ضروری است. در مراحل اولیه تکوین رویان، این مسیر باعث مهاجرت سلولهای ستیغ عصبی قلبی به مکان اولیه تشکیل قلب، یعنی مجرای خروجی قلب (cardiac outflow tract) میشود، بگونهای که سلولهایی که سیگنال SHH را دریافت میکنند، فاکتور رونویسی قلبی GATA4 را در ناحیه ثانویه قلبی یا second heart field (SHF) فعال میسازند (60). پروتئین BMP که در مسیر ابرخانواده سیتوکاینهای TGF-β عمل میکند، از طریق فعالسازی فاکتور رونویسی Smad، نقش ضروری در تشکیل صفحه اولیه قلبی یا first heart field (FHF) ایفا میکند (61). رتینوئیک اسید نیز با محدودکردن رشد در ناحیهی ثانویه قلبی، در تنظیم تکوین قلب مشارکت دارد. جهش در ژن Raldh2، که مسئول سنتز رتینوئیک اسید است، این محدودیت رشدی را از بین میبرد، در نتیجه، بخشهایی از قلب که از ناحیهی ثانویه قلبی منشأ میگیرند، با نقصهای ساختاری مواجه خواهند شد (64-62). شکل ۲-ب مسیر پیامرسانی دارای نقش عملکردی مهم در هر مرحله از تکوین قلب رویان را نمایش میدهد.
نقش عوامل اپیژنتیک (Epigenetics) در تکوین قلب: پژوهشهای متعددی نشان دادهاند که تغییرات اپیژنتیک (epigenetic modification) از طریق مکانیسمهایی مانند متیلاسیون DNA (DNAmethylation)، نوسازی یا رمدلینگ ساختار کروماتین (Chromatin remodeling)، تغییرات هیستونی (Histone modifications) و تنظیم بیان ژنها (gene expression regulation) توسط RNAهای غیرکدکنندهی کوچک (MicroRNA) و بزرگ Long-noncoding RNAs))، بدون تغییر در توالی نوکلئوتیدی DNA، بیان ژنهای دخیل در تکوین قلب را کنترل میکنند و نقشی کلیدی در ایجاد CHD دارند (70-65، 57). متیلاسیونDNA (DNA methylation): متیلاسیون DNA، یکی از نخستین مکانیسمهای کشفشده اپیژنتیکی است که شامل افزودن گروه متیل به نوکلئوتیدهای سیتوزین در جزایر CpG (CpG islands) است و در فرایندهای بیولوژیکی مختلف از جمله تکوین قلب نقش دارد. در طی رشد رویان، بلوغ کاردیومیوسیتها با تغییرات متیلاسیون DNA همراه است. ابتدا، یک موج دمتیلاسیون (demethylation) در ژنهای قلبی، بهویژه ژنهایی که پروتئینهای سارکومر (sarcomere) را کد میکنند، رخ میدهد و پس از تولد نوزاد، روند متیلاسیون مجدد، شکل میگیرد. هرگونه تغییر غیرطبیعی در متیلاسیون DNA میتواند از طریق تأثیر بر بیان ژن، به بروز CHD منجر شود (71). در این فرآیند، فولات (Folate) یا ویتامین B9 بهعنوان منبع اصلی گروههای متیل برای متیلاسیون DNA، نقشی حیاتی دارد. مطالعات نشان دادهاند که مصرف مکمل فولات توسط مادران میتواند خطر ایجاد CHD را در نوزادان مبتلا به سندرم داون (Down syndrome) کاهش دهد. آنزیم متیلنتتراهیدروفولات ردوکتاز یا Methylenetetrahydrofolate reductase (MTHFR) که در متابولیسم فولات نقش کلیدی دارد، در صورت کاهش فعالیت میتواند منجر به ایجاد شکستگی در DNA و تفرق غیرطبیعی کروموزومها شود. پژوهشها نشان دادهاند که افزایش متیلاسیون پروموتر MTHFR در مادران دارای فرزندان مبتلا به CHD و سندرم داون بهطور قابلتوجهی افزایش یافته است. همچنین، در کودکان مبتلا به CHD، سطح بالاتری از نشانگرهای زیستی متیلاسیون مشاهده شده است که این افزایش با ناهنجاریهای پیچیدهی قلبی در ارتباط است (72).
نوسازی ساختار کروماتین (Chromatin remodeling): نوسازی (رمدلینگ) ساختار کروماتین یکی دیگر از مکانیسمهای مهم اپیژنتیک است که وابسته به فعالیت چهار کمپلکس مرتبط با ATP میباشد: کمپلکس SWI/SNF (SWItch/Sucrose Non-Fermentable)، پروتئین Chromodomain helicase DNA-binding (CHD) ، پروتئین Iswi و پروتئینINO80 . حذف پروتئین BRG1 (یک فعالکننده رونویسی کد شده توسط ژن SMARCA4)، یکی از اجزای کمپلکس BRG-/BRM-associated factor (BAF) که همولوگ کمپلکس SWI/SNF در مهرهداران است، موجب نقص در تشکیل دیواره بین بطنی و همچنین ناهنجاریهایی در بطن راست و مجرای خروجی قلب میشود. کاهش مقدار Baf60c، یکی دیگر از اجزای کمپلکس BAF، در موشها منجر به نقص در نواحی قدامی قلب، اختلال در خمیدگی لوله قلبی در طی تکوین جنین، کوتاهی مجرای خروجی قلب و هیپوپلازی بطن میشود. علاوه بر این، ناکفایتی هاپلوئیدی ژن CHD7 با سندرم CHARGE مرتبط است که در مبتلایان آن، نقصهای دیواره بین دهلیزی، دیوارهی بین بطنی، دیواره دهلیزی-بطنی و ناهنجاریهای کونوتراکال قلب (conotruncal heart defects) دیده میشود (73,74).
تغییرات شیمیایی هیستونها (Histone modifications): مکانیسمهای تغییرات هیستونی شامل اثر هیستونداستیلازها یا Histone deacetylases (HDACs)، هیستوناستیلازها یا Histone acetyltransferases (HATs) و هیستونمتیلترنسفرازها یاHistone methyltransferases (HMT) بر DNA میشوند. حذف پروتئینهای HDAC5 و HDAC9 در سلولهای رده زایا با نقص دیواره بین بطنی مرتبط است (75). حذف SMYD1 که یک متیل ترانسفراز هیستونی است موجب هیپوپلازی بطن میشود. حذف WHSC1، یک متیل ترانسفراز هیستونی دیگر، با نقصهای دیواره بین دهلیزی و دیواره بین بطنی مرتبط است. همچنین، حذف هیستوندمتیلاز در سلولهای رده زایا با ناهنجاریهای پیچیدهای، مانند خروجی دوگانه بطن راست و هایپرتربکولاسیون (hyper trabeculation) همراه است (76). نواحی غیر کدکننده (Non-coding) ژنوم: تنها 1 تا 2 درصد از DNA ژنومی انسان از اصل «Central dogma» پیروی میکند، به این معنی که DNA به RNA رونویسی میشود و سپس به پروتئین ترجمه میشود. حدود ۹۸ درصد دیگر، کدکننده پروتئین نیستند که به آن DNA غیرکدکننده (Non-coding DNA) نیز گفته میشود. حدود ۲۰ درصد از این DNA که پیش از این از نظر عملکردی «بیفایده یا junk» خوانده میشدند، در میان مهرهداران مختلف بسیار حفاظت شدهاند و حدود 80 درصد آنها عملکردهای بیوشیمیایی مهمی در سلول دارند (77). RNAهای غیرکدکننده در سالهای اخیر بهعنوان عوامل کلیدی در افزایش خطر ابتلا به انواع بیماریهای انسانی، از جمله بیماریهای مادرزادی قلب، شناسایی شدهاند. این مولکولها با تنظیم فاکتورهای رونویسی، تکثیر کاردیومیوسیتها، و همچنین تمایز و تکوین عضلات قلبی و اسکلتی، نقش مهمی در فرآیندهای زیستی ایفا میکنند (81-78). بخش عظیمی از تحقیقات کنونی، در حال تلاش برای شناسایی عملکردهای بیوشیمیایی جدید برای DNAهای غیرکدکننده است، تا دادههای حاصل از تحقیقات را به فنوتیپهای مولکولی، فیزیولوژیکی و پاتولوژیکی مرتبط کند. تا پیش از این، به دلیل کمبود دادههای ژنوم انسان و عدم وجود الگوریتمها و روشهایی برای ارتباط DNA نادر یا نوظهور با عملکرد و فنوتیپ بیماران، بیماریزایی واریانتهای غیرکدکننده قابل بررسی نبود. با اینحال، برخی از مطالعات، شواهدی ارائه میدهند که بخشهای غیرکدکننده ژنوم، برای تکوین قلبی-عروقی ضروری هستند (82). بهعنوان مثال، واریانتهای نادر هموزیگوت غیرکدکننده در سندرم هولت-اورام، با ایجاد اختلال در عناصر تنظیمکننده اختصاصی قلب، باعث بروز CHDهای ایزوله میشوند (83). همچنین شواهدی وجود دارد که نشان میدهند واریانتهای نادر غیرکدکننده نوظهور در نزدیکی ژنهای شناختهشده مرتبط با CHD، در گروههای بیماران، نسبت به گروه کنترل، بیشتر مشاهده میشوند و حتی زیرمجموعهای از این واریانتهای غیرکدکننده نوظهور، بیان ژنهای CHD را نیز تغییر میدهند )84(. اگرچه مطالعات همراهی در سطح ژنوم، از قدرت آماری محدودی برخوردارند، اما تاکنون، واریانتهای غیرکدکنندهی شایع در ارتباط با بیماریهای قلبی مادرزادی، بهویژه در نقصهای دیوارهای و نقصهای انسدادی مجرای خروجی بطن چپ شناسایی شدهاند (87-85). اخیراً، یک متا- آنالیز که چهار گروه CHD با 55,342 شرکتکننده را با پنل مرجع TOPMed مقایسه کرده است، 16 موقعیت کروموزومی مرتبط با خطر CHD را شناسایی کرد (88(. از این 16 موقعیت، 13 مورد به ژنهای مرتبط با تکوین قلبی مربوط میشوند. این یافتهها بر اهمیت بیشتر بررسی نقش عملکردی ژنوم غیرکدکننده و درک بهتر ارتباط آن با استعداد ابتلا به CHD تأکید میکنند (89). MicroRNAها (miRNA)و نقش آنها در بروز CHDMicroRNAها: مولکولهای کوچک RNA غیرکدکنندهای هستند (با طول ۱۹ تا ۲۲ نوکلئوتید) که بیان ژن در یوکاریوتها را در سطح پس از رونویسی تنظیم میکنند. این مولکولها در تعامل با ناحیه 3'UTR از RNA پیامرسان (mRNA) ژن هدف، مانع از بیان آن ژن میشوند (91, 90). تولید میکروRNAها در انسان، با رونویسی یک میکروRNA اولیه (pri-miRNA) توسط RNA پلیمراز II آغاز میشود که حاوی ساختاری سنجاقمانند است. این ساختار توسط کمپلکس DROSHA-DGCR8 به یک پیشساز میکروRNA (pri-miRNA) ۶۰ تا 90 نوکلئوتیدی با انتهای چسبنده در 3'UTR پردازش میشود. این پیشساز میکروRNA از طریق پروتئین XPO5 در فرایندی وابسته به RAN-GTP به سیتوپلاسم منتقل میشود، جاییکه توسط DICER (RNase III endoribonucleases) و Transactivation response RNA binding protein (TRBP) به یک دورشتهای 19 تا 22 نوکلئوتیدی تبدیل میشود. نهایتا یک رشته، بر اساس محتوای پورین و پایداری ترمودینامیکی انتهای 5'UTR، انتخاب شده و در کمپلکس RNA-induced silencing complex (RISC) ادغام میشود تا تخریب، ناپایداری یا مهار ترجمه mRNA هدف را هدایت کند (92). برهمکنش miRNA و هدف آن توسط ناحیهی seed region ( شامل نوکلئوتیدهای ۲ تا ۸ در 5'UTR میکروRNA) میانجیگری میشود. محلهای اتصال مکمل و معکوس میکروRNA (Reverse-complementary miRNA binding sites) که بهعنوان عناصر پاسخدهنده به میکروRNA (MREs) نیز شناخته میشوند، معمولاً در نواحی 3'UTR از RNAهای پیامرسان (mRNA) هدف قرار دارند، اما میتوانند در نواحی 5'UTR یا توالی کدکننده پروتئین نیز وجود داشته باشند (92). برخی miRNAها در بین گونههای متنوع جانوری حفاظت شدهاند و در بافتهای خاصی بیان میشوند. این بیان اختصاصی بافتی miRNAها، در رشد، تکثیر و تمایز بافتها، تصمیمگیری برای سرنوشت ردههای سلولی و همچنین homeostasis بافت، اثرگذار است. از میان miRNAهایی که به طور خاص در بافت قلب بیان میشوند، miR-1 به عنوان فراوانترین miRNA در قلب جوندگان شناسایی شده است (حدود 45% از کلmiRNAهای قلبی را شامل میشود). miR-1b، miR-1d، miR-133، miR-206، miR-143 و miR-208، شش miRNA با بیان اختصاصی در عضله اسکلتی و قلب هستند که از بررسی بیان ۱۱۹ میکروRNA در اندامهای انسان و موش یافت شدند (94, 93). در این میان، نقش miR-1، miR-133، و miR-208b در تکوین رویانی قلب انسان تائید شده است (97-95). این miRNAهای اختصاصی عضله، به عنوان MyomiRs نامگذاری شدند و نقش اساسی در تکوین قلب ایفا میکنند (98). برخی از این miRNAها در شکل ۳ در کنار هر مرحلهی تکوینی نمایش داده شدهاند. هرگونه اختلال و جهش جدید در محل اتصال RNAهای غیر کدکننده روی DNA که در نقش و عملکرد تنظیمی آنها تغییری ایجاد کند، میتواند باعث افزایش استعداد ابتلا به CHD شود. بنابراین مطالعۀ نواحی نظیر 3'UTR ژنهای هدف، میتواند یکی از راههای مهم تشخیص مکانیسم پاتوژنز این بیماری و حائز اهمیت مطالعه بیشتر محققان باشد (99،100).
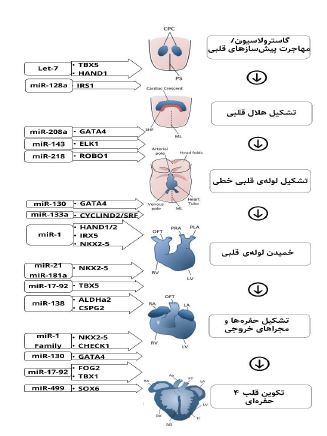
شکل ۳. نقش miRNAها در تنظیم مراحل مختلف رشد قلب. فلشها بیانگر تنظیم فاکتور رونویسی توسط miRNA مربوطه میباشد.
اختصارات: Ao: آئورت، CPC: سلولهای پیشساز قلبی، IVS: سپتوم بینبطنی، L: چپ، LA: دهلیز چپ، LV: بطن چپ، ML: خط میانی، OFT: مسیر خروجی، P: خلفی، FHF: ناحیه اولیه قلبی، PLA: دهلیز چپ اولیه، PRA: دهلیز راست اولیه، PS: خط اولیه، PT: تنه ریوی، R: راست، RA: دهلیز راست، RV: بطن راست، SHF: ناحیه ثانویه قلبی، Tr: ترابکولا (102, 101).
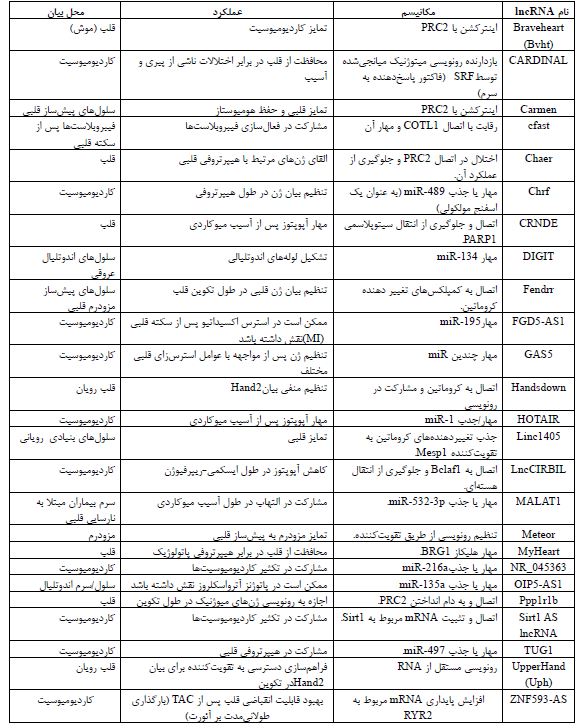
lncRNAها و نقش آنها در بروز CHD: RNAهایی که طولشان بیشتر از 200 نوکلئوتید بوده و به هیچ محصول پروتئینی ترجمه نمیشوند، در یک دسته به نام RNAهای غیر کدکننده بلند Long-noncoding RNAs)) یا lncRNAها گروهبندی میشوند. lncRNAها معمولاً با تنظیمات پیرایشی، برش داده میشوند، یک کلاهک در جهت 5'UTR و دم poly(A) در جهت 3'UTRدارند و الگوهای بیانی وابسته به مکان و زمان در بافتهای متنوع، از جمله بافت قلب از خود نشان میدهند. برخلاف mRNAها، توالی اکثر lncRNAها، بهطور میانگین حفاظتشدگی (conservation) ضعیفی در میان گونههای جانوری دارند که این امر تردیدهای اولیهای را در مورد اهمیت عملکردی آنها، در کنترل فرآیندهای زیستی اساسی، مانند تکوین قلب بهوجود میآورد. در واقع، بهنظر میرسد بیشترlncRNAها، مختص گونه هستند. بهعنوان مثال، بهنظر نمیرسد که تقریباً 70 درصد از lncRNAهای انسانی، همولوگی با توالی مشابه در هیچ گونهی دیگری داشته باشند. از یک سو، این علت میتواند توانایی تحقیق و بررسی عملکردی lncRNAهای خاص انسان را در مدلهای غیرانسانی محدود کند و از سوی دیگر، نشان میدهد که lncRNAهای غیرانسانی، به دلیل عدم حضور توالی مشابه آنها در ژنوم انسان، برای بروز بیماری در انسان ضروری نیستند. با اینحال، بیش از هزار همولوگ lncRNA انسانی احتمالی در سایر گونهها شناسایی شده است که نشان میدهد بسیاری ازlncRNAها ممکن است برای تنظیم فرآیندهای زیستی حفاظتشدهای اهمیت داشته باشند. در واقع، مطالعات in vivo که در سالهای اخیر بر روی lncRNAها انجام شدهاند، هیچ شکی باقی نگذاشتهاند که lncRNAها برای سلامت قلبی- عروقی جنین، از اهمیت زیادی برخوردارند، بهطوری که نقش چندین lncRNA برای تکوین قلب به اندازهی نقش پروتئینهای حیاتی قلبی ضروری است. همانند سایر بافتها، lncRNAها در تمام انواع سلولهای قلب، از جمله کاردیومیوسیتها، سلولهای اندوتلیال، سلولهای عضله صاف عروقی و فیبروبلاستها یافت میشوند. lncRNAها در تمام مراحل تکوین قلب بیان میشوند و بهطور دینامیک و پویایی تنظیم میشوند. lncRNAها بهعنوان کمک-تنظیمکنندههای (co-regulators) شبکههای رونویسی و مسیرهای پیامرسانی، متشکل از پروتئینها و RNAهای غیرکدکننده دیگر، مشارکت دارند و همراه با اجزای دیگر این شبکه تنظیمی، الگوی دقیق بیان ژنهای مورد نیاز برای تکوین طبیعی قلب در موقعیت و زمانهای مشخص را هماهنگ میکنند. هرگونه اختلال در این شبکههای تنظیمی، میتواند تاثیرات ویرانگری بر تکوین اولیهی قلب داشته باشد. اختلال در lncRNAهای قلبی در طول مورفوژنز اولیه قلب، میتواند منجر به نقص در تعهد ردههای سلولی قلبی (cell lineage commitment)، روند تشکیل بطن و جداشدن صحیح دیوارهها در قلب شود (104, 103). بهطور کلی، ایجاد پیشساز miRNA، پایدارکنندگی ساختار miRNA، رقابت با miRNA برای تنظیم رونویسی، جذب miRNA و اتصال به آن و در نتیجه مهار کردن فعالیت تنظیمی miRNA، ایجاد داربست و برهمکنش با پروتئین و DNA و RNA (تشکیل کمپلکس ریبونوکلئوپروتئین)، تشکیل ساختارهای تریپلکس (triplex) با اتصال به دو رشتهای DNA و عمل به عنوان راهنمای جابجایی فاکتورهای تنظیمی در سلول، از جمله نقشهای تنظیمی lncRNAها هستند. همچنین در حال حاضر، بسیاری از lncRNAهای تنظیمکننده تکوین، تکثیر و تمایز بافتی، با منشأ مزودرمی یافت شدهاند که از جهت مطالعۀ روند ایجاد بیماری، به عنوان بیومارکرهای مولکولی قابل توجه هستند (106, 105). در جدول 6، شماری از lncRNAهای اثرگذار در تکوین قلب به همراه نقش آنها نام برده شده است و در شکل ۴ بخشی از عملکردهای سلولی lncRNA به تصویر کشیده شده است (103).
جدول۶: مشخصات برخی از lncRNAهای مرتبط با تکوین قلب با عملکردهای شناسایی شده.
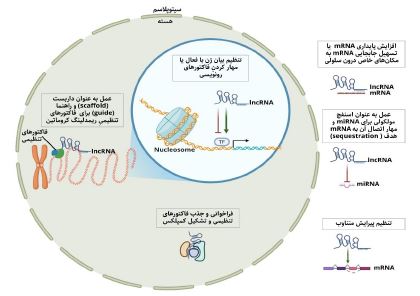
شکل ۴. انواع نقشهای عملکردی lncRNA درسلول. lncRNA در تعامل با DNA، پروتئین، miRNA و mRNA انواع نقشهای تنظیمی را ایفا میکند.
نتیجهگیری
مطالعه حاضر، دیدگاهی جامع در مورد عوامل ژنتیکی و اپیژنتیکی مؤثر بر تکوین قلب و بیماریهای مادرزادی قلب (CHD) ارائه داده است. پیشرفتهای اخیر در ویرایش ژنوم و بهرهگیری از قابلیتهای سلولهای بنیادی پرتوان، امکان شناسایی دقیق عملکرد ژنهای اثرگذار بر تکوین قلب را فراهم کردهاند. با اینکه هنوز تمام جنبههای بیماریزایی ژنهای مرتبط با CHD بهطور کامل شناخته نشده است، روشهای جدید تشخیصی و بهبود طراحی آزمایشها، نویدبخش یافتن مکانیسمهای مولکولی کلیدی در ایجاد این بیماری هستند. این تحقیقات بینشی جامع پیرامون فرآیندهای مرتبط با تکوین قلب در اختیار متخصصان، محققان، پزشکان و سیاستگذاران حوزه سلامت خواهد گذاشت. همچنین، این یافتهها در کنترل، مدیریت، پیشبینی و درمان CHD مؤثر خواهد بود و موجب ارتقای سلامت مبتلایان خواهد شد.
سپاسگزاری
نویسندگان مقاله از حمایتهای دانشگاه یزد در انجام این پژوهش که در راستای پایاننامه کارشناسی ارشد خانم تبریزی بوده است، تشکر و قدرانی میکنند.
حامی مالی: ندارد.
تعارض در منافع: وجود ندارد.
مشارکت نویسندگان
در ایده، نگارش و ویرایش مقاله کلیه نویسندگان مشارکت داشتند.
بیماری مادرزادی قلب یا Congenital Heart Disease (CHD) بهعنوان یک ناهنجاری آناتومیکی شدید در بافت قلب یا عروق بزرگ مرتبط با آن تعریف میشود که از لحاظ اثر بر عملکرد طبیعی بدن اهمیت دارد. CHD شایعترین ناهنجاری مادرزادی در سراسر جهان است که حدود 1 درصد از تولدهای زنده را تحت تأثیر قرار میدهد. این بیماری که علت اصلی مرگومیر ناشی از نقصهای مادرزادی است، منجر به سقط حدود 10 درصد از جنینها در دوران بارداری میشود. بهطور جهانی، تقریباً حدود ۲/۱ میلیون نفر در سال، با نقصهای گسترده قلبی متولد میشوند (1,2). علل زمینهای بروز CHD نسبتاً کمتر شناخته شده است و ترکیبی از عوامل ژنتیکی، اپیژنتیکی و محیطی در ایجاد آن نقش دارند. شواهد اپیدمیولوژیک بر تأثیر غالب عوامل ژنتیکی تأکید دارند، از جمله افزایش خطر ابتلا در افرادی با سابقه خانوادگی، تطابق بیشتر در دوقلوهای همسان نسبت به غیرهمسان، و شیوع بالاتر در جمعیتهایی با ازدواجهای فامیلی (6-3). با اینحال، درصد قابلتوجهی از موارد بیماری، در خانوادههای بدون سابقه قبلی از CHD رخ میدهد که نشاندهنده وقوع جهشهای جدید (de novo)، مانند ناهنجاریهای کروموزومی (8%-12%)، واریانتهای تعداد نسخه ژنی یا Copy Number Variant (CNV) (3%-25%) و جهشهای نقطهای (3%-5%) است (4). در مقابل، عوامل غیرژنتیکی شناختهشده، شامل تراتوژنهای محیطی، مواجهات مادر با عوامل خطر در دوران بارداری و عوامل عفونی که 8%-12% کل موارد بیماران را تشکیل میدهند (7). تخمین زده میشود در حدود 400 ژن در پاتوژنز CHD نقش داشته باشند. جهش در ژنهای کدکننده پروتئینهای مرتبط با بازسازی (رمدلینگ) کروماتین، سیگنالینگ سلولی و فاکتورهای رونویسی میتوانند فرآیند تمایز سلولهای قلبی را مختل کرده و به اختلالات ساختاری و عملکردی قلب منجر شوند (8). با اینحال، علل ژنتیکی CHD، در تقریبا 80% موارد ناشناخته است. این بیماری به دلیل تنوع ژنتیکی و هتروژنسیتی پیچیده (حالتی که جهشهای ژنی مختلفی باعث بیماری میشوند)، غالبا الگوهای وراثتی مندلی را دنبال نمیکند. این هتروژنسیتی منجر به درصد نفوذ و بیان متغیر ژنهای دخیل میشود (11-9). اگرچه ژنهای کاندید متعددی برای بروز CHD سندرمی شناسایی شدهاند، بررسی حالات CHD غیرسندرمی، به دلیل هتروژن بودن آن چالشبرانگیزتر است. امروزه پیشرفتهای چشمگیر در تکنولوژیهای توالییابی DNA، شناسایی واریانتهای نادر در ژنهای جدید مرتبط با CHD غیرسندرمی را نیز ممکن ساخته است (15-12). تکنیکهایی مانند توالییابی نسل جدید یا Next-generation sequencing (NGS)، توالییابی RNA تکسلولی (Single Cell RNA sequencing)، توالییابی microRNA (miRNA sequencing) و آنالیز سلولهای بنیادی پرتوان القایی انسانی (hiPSCs) Human-Induced Pluripotent Stem Cell در شناسایی علل و سازوکارهای بیماری CHD بسیار مؤثر بودهاند (16-18). علاوه براین، مطالعه و بررسی بیماریزا بودن واریانتهای جدید و با اهمیت نامشخص یا Variants of Uncertain Significance (VUS) با استفاده از سلولهای iPSC ویرایششده توسط CRISPR امکانپذیر شده است (19). این تکنیکها با ارزیابی نقایص عملکردی واریانتهای خاص، کیفیت زندگی نوزادان مبتلا به CHD را بهبود بخشیدهاند، بهطوری که بیش از 90% آنها به سنین بزرگسالی میرسند (20). مطالعات مختلفی برای بررسی علتشناسی بیماریهای مادرزادی قلب انجام شده است. با اینحال، شناسایی عوامل مولکولی و مکانیسمهای مرتبط با این بیماری، همچنان موضوع بحث و تحقیقات متخصصین است. درک علل ژنتیکی منجر شونده به CHD میتواند بینش عمیقی در مورد زیربنای مولکولی این بیماری فراهم کند و در تعریف تخمین ریسک بیماری و بهبود روشهای پیشگیری از آن مؤثر باشد. همچنین، چنین دانشی میتواند برنامهریزی برای درمانهای نوین را تسهیل کند و اساس زیستشناسی تکوین قلب را روشنتر نماید و قادر است به پیشبینی نتایج بالینی، ارزیابی خطر در خانوادهها و غربالگری افراد در معرض عوامل خطر کمک کند. علاوه براین، مطالعهی ارتباطات ژنوتیپ-فنوتیپ در این بیماری، میتواند اطلاعات ارزشمندی درباره پیشآگهی (prognosis) بیماری CHD ارائه دهد. این مقاله مروری به جدیدترین یافتهها درباره زیربنای ژنتیکی این بیماری میپردازد.
روش بررسی
برای دستیابی به نتایج این پژوهش مروری، نخستین گام یک جستجوی جامع در پایگاههای دادهای معتبر علمی، از جمله PubMed، Scopus و ScienceDirect بود. در این مرحله، با استفاده از کلیدواژههای تخصصی مرتبط با موضوع مانند «congenital heart defect»،«congenital heart disease»، «genetic basis»،«epigenetic modifications»، «molecular mechanisms»، و دیگر عبارات جستجوهایی طراحی و اجرا شدند تا متون علمی مرتبط شناسایی گردد. این جستجوها بهگونهای انجام شدند که هم مطالعات قدیمیتر و هم پژوهشهای منتشرشده در سالهای اخیر مورد بررسی قرار گیرند و بدین وسیله، تصویر جامع و بهروزی از مبنای ژنتیکی و اپیژنتیکی CHD ارائه شود. پس از شناسایی مقالات، گزینش آنها بر اساس معیارهای پیشتعیینشده صورت گرفت. از جمله این معیارها میتوان به جدیدبودن یافتهها (انتشار در سالهای اخیر)، وجود اعتبار علمی (داشتن روند داوری علمی و انتشار در نشریات معتبر) و پوشش موضوع اشاره کرد. مقالاتی که معیارهای مورد اشاره را نداشتند، از انتخاب حذف شدند. در مرحله بعد، اطلاعات کلیدی از ۸۶ مقاله منتخب استخراج گردید. این اطلاعات شامل یافتههای مهم در زمینههای مختلف از جمله واریانتهای ژنتیکی، تغییرات متنوع و متعدد اپیژنتیکی و مکانیسمهای مولکولی مربوط به CHD بود. فرآیند استخراج دادهها به دقت و بهصورت دستی انجام شد تا از صحت و دقت اطلاعات اطمینان حاصل شود. سپس، دادههای استخراجشده دستهبندی و تحلیل شدند. این تحلیل شامل بررسی نقاط مشترک و تفاوتهای میان مطالعات و تبیین ارتباط بین یافتههای بهدستآمده بود. هدف از دستهبندی فاکتورهای ژنتیکی و اپیژنتیکی مورد بررسی، ارائه یک دیدگاه جامع و انتقادی نسبت به همه فاکتورهای ژنتیکی و اپیژنتیکی دخیل در ایجاد CHD بود که به انعکاس وضعیت دانش موجود در این حوزه، و همچنین خلأهای پژوهشی بپردازد و به مطرحشدن پیشنهادهایی در مورد موضوع جهتگیریهای تحقیقات آتی کمک نماید. در نهایت، دادههای طبقهبندی شده در قالب یک دیدگاه یکپارچه و علمی ارائه شدند.
آمارهای جمعیتی و دستهبندی بیماریهای مادرزادی قلبی: بیماریهای مادرزادی قلب یا Congenital Heart Disease (CHD) یک طیف گسترده از ناهنجاریهای ساختاری قلب هستند که در دوران رشد رویان ایجاد میشوند و از عوامل مهم در بروز بیماری و مرگ و میر در کودکان هستند. با وجود پیشرفتهای عظیم در تشخیص و مدیریت بیماریهای قلبی مادرزادی در چند دهه گذشته، بیشتر اطلاعات موجود درباره مدیریت بیماریهای قلبی مادرزادی از مناطقی با مقدار بالای شاخص اجتماعی- جمعیتشناختی یا Socio-demographic Index (SDI) بهدست آمده است. با وجود محدودیت جمعآوری داده، از کشورهای با SDI پائین، «آنالیز سیستماتیک بیماریهای مادرزادی قلب- Global Burden of Disease (GBD) 2017» جامعترین ارزیابی جهانی از آمار بیماریهای مادرزادی قلب تا به امروز را ارائه میدهد. این آنالیزها که حاصل همکاری پژوهشی چندین مرکز تحقیقاتی هستند، توسط مؤسسه Institute for Health Metrics and Evaluation (IHME) تجمیع و در سال ۲۰۲۰ منتشر شده است. این دادهها توسط متخصصان، پزشکان، آموزشدهندگان، محققان و سیاستگذاران در سراسر جهان برای کنترل بیماری CHD، تدوین سیاستهای بهداشتی مؤثر و گرفتن تصمیمات کلیدی در رابطه با بهبود زندگی بیماران استفاده میشود (21). گزارش GBD به ارزیابی بروز و مرگومیر بیماری CHD، آسیبهای ناشی از بیماری و عوامل خطر بیماریها در سطح جهانی میپردازد. طبق دادههای اپیدمیولوژیک ارائه شده، تخمین زده شده است که در سال ۲۰۱۷ تقریباً 12 میلیون نفر در جهان مبتلا به بیماریهای مادرزادی قلب بودهاند که نشاندهنده افزایش ۷/۱۸ درصدی نسبت به سال 1990 میباشد. تعداد مرگومیرهای مرتبط با بیماری در سال 2017 حدود ۲۶۱ هزار نفر برآورد شده است که کاهش ۵/۳۴ درصدی نسبت به تعداد تخمین زده شده در سال 1990 دارد. بیماریهای مادرزادی قلبی، بهعنوان یکی از علل اصلی مرگومیر نوزادان شناخته میشوند که در سطح جهانی رتبه ششم و در مناطق با SDI بالا، رتبه دوم را دارا هستند. با تغییر علل اصلی مرگومیر از بیماریهای واگیردار به بیماریهای غیرواگیردار، انتظار میرود اهمیت بیماریهای قلبی مادرزادی، به عنوان یکی از عوامل اصلی مرگومیر نوزادان در سطح جهانی در سالهای آینده بیشتر مورد توجه قرار گیرد (21). شکل ۱ جدیدترین آمار میزان مرگومیر ناشی از بیماریهای مادرزادی قلبی در هر ۱۰۰۰۰۰ نفر طبق گزارش GBD سال ۲۰۲۰ در سراسر جهان به تفکیک هر کشور را نمایش میدهد. سیستم کدگذاری بینالمللی برای بیماریهای مادرزادی قلب یا International Paediatric and Congenital Cardiac Code (IPCCC) که شامل۳۶۷ عنوان بیماری استاندارد است، یک چارچوب طبقهبندی شده برای این ناهنجاریها ارائه میدهد. این سیستم توسط انجمن بینالمللی نامگذاری و سازمان بهداشت جهانی، در ویرایش یازدهم از «دستهبندی جهانی طبقهبندی بیماریها» یا International Classification of Diseases 11th Revision (ICD-11) معرفی شده است. طبق این طبقهبندی استاندارد، بیماریهای مادرزادی قلب بهطور کلی به چهار دسته اصلی تقسیم میشوند. نقایص هیپوپلازی (Hypoplasia defects) یکی از این دستههاست که در آن تکامل یک طرف از قلب ناقص میماند و توانایی پمپاژ خون در آن سمت مختل میشود. بسته به سمت درگیر، این ناهنجاری به دو شکل اصلی، شامل سندرم قلب چپ هیپوپلاستیک یا Hypoplastic Left Heart Syndrome (HLHS) و سندرم قلب راست هیپوپلاستیک یا Hypoplastic Right Heart Syndrome (HRHS) دیده میشود. دسته دوم، نقایص انسدادی یا Obstructive Heart Defects (OHDs)، شامل ناهنجاریهایی است که مسیر جریان خون در دریچهها، سیاهرگها یا سرخرگها را تنگ یا مسدود میکنند. از جمله نمونههای این دسته، میتوان به کوارکتاسیون آئورت یا Coarctation of Aorta (CoA) و تنگی آئورت یا Aortic Stenosis (AS) اشاره کرد. دسته سوم، نقایص دیوارهای (Septal Defect)، شایعترین نوع بیماریهای مادرزادی قلب هستند. این ناهنجاریها به دلیل نقص یا سوراخ در دیوارههای قلب ایجاد میشوند که منجر به اختلال در جریان طبیعی خون بین حفرههای قلب میشوند. نقص دیواره بین بطنی یا Ventricular septal defect (VSD) و نقص دیواره بین دهلیزی یا Atrial Septal Defect (ASD) از جمله نمونههای رایج این دسته هستند. در نهایت، نقایص سیانوتیک یا Cyanotic Heart Disease به ناهنجاریهایی گفته میشود که باعث کاهش اکسیژنرسانی به بافتها شده و معمولاً با کبودی (سیانوز) همراه هستند. این دسته شامل مواردی مانند اتصال نابجای همه وریدهای ریوی یا Total Anomalous Pulmonary Venous Connection (TAPVC)، تترالوژی فالو یا Tetralogy of Fallot (ToF)، جابجایی سرخرگهای بزرگ یا Transposition of the Great Arteries (TGA) و تنه مشترک سرخرگی پایا یاPersistent Truncus Arteriosus (PTA) است. جدول ۱ شماری از شایعترین انواع بیماریهای مادرزادی قلب در نوزادان را نمایش میدهد. طبقهبندی دقیق این بیماریها کمک میکند تا پزشکان ناهنجاریهای قلبی مادرزادی را بهتر تشخیص داده و درمان مناسب را برنامهریزی کنند. علاوه بر این، این دستهبندی استاندارد به جمعآوری دادههای دقیق برای تحقیقات و پیشگیری کمک شایانی میکند (22).
عوامل ژنتیکی موثر در بروز CHD: پیشرفت در تکنیکهای مولکولی، امکان مطالعه نقصهای تکوینی قلب را فراهم کرده و دانش محققین را از مورفولوژی بیماری و عوامل ژنتیکی دخیل در آن افزایش داده است. شواهد بسیاری نشان میدهند که عوامل ژنتیکی، نقش کلیدی در پاتوژنز CHD ایفا میکنند. جهشهای نقطهای در ژنهای فاکتورهای رونویسی قلب، پلیمورفیسمهای تکنوکلئوتیدی یا Single-Nucleotide Polymorphism (SNP)، آنیوپلوئیدیهای کروموزومی (Chromosomal aneuploidies)، واریانتهای تعداد نسخه کروموزومی یا Copy Number Variation (CNV) و جهش در ژنهای گیرندهها و لیگاندهای مربوط به مسیرهای پیامرسانی مورفوژنز قلب، همگی از عوامل ژنتیکی مرتبط با بروز CHD هستند. جهشها و تغییرات ژنتیکی در این عوامل رونویسی، با بسیاری از CHDهای غیرسندرمی مرتبط است. مطالعات عملکردی بر روی این ژنها در آزمایشات حیوانی نشاندهنده نتایج قابلاعتماد و تکرارپذیری بوده است که مدل توارث تکژنی برای پاتوژنز CHD را پیشنهاد میدهند. با اینحال، مدل توارث تکژنی دو سؤال کلیدی و مهم را مطرح میکند: اول آنکه، چرا انواع مختلفی از CHD با یک نوع جهش ژنی دیده میشوند؟ دوم آنکه، چرا جهشهای متفاوت تکژنی باعث ایجاد فنوتیپهای مشابه CHD میشوند؟ این پرسشها نشان میدهند که احتمالاً عوامل متعدد و مدل توارث چندعاملی در علتشناسی CHD دخیل هستند (23). یکی از جنبههای جالب توجه CHD این است که با وجود کاهش پتانسیل تولیدمثلی در بیماران مبتلا، شیوع این بیماری در جمعیتها ثابت باقی مانده است. این امر احتمال رخداد جهشهای نوظهور را مطرح میکند. با اینحال، انتظار میرود که روند انتخاب طبیعی، باعث کاهش شیوع CHD شود، زیرا این بیماریها با میزان مرگومیر بالا و کاهش توان تولیدمثل مرتبط هستند. همچنین، هنوز مشخص نیست که CHD دارای الگوی توارث مشخصی باشد و اینکه آیا بهصورت غالب اتوزومی و یا مغلوب اتوزومی به ارث میرسد. جهشهای غالب اتوزومی معمولاً با نفوذ بالایی همراه هستند و انتظار میرود درصد زیادی از بستگان درجهیک، CHD را به ارث ببرند. با اینحال، مشاهدات نشان میدهند که این فرضیه همیشه صحیح نیست. بهنظر میرسد الگوی مغلوب اتوزومی برای توضیح اساس ژنتیکی CHD مناسبتر باشد. با توجه به هتروژن بودن این بیماری، مطالعات همراهی سراسر ژنوم یا Genome-wide association studies (GWAS) ابزار ارزشمندی برای شناسایی عوامل ژنتیکی متعددی است که به بروز CHD کمک میکنند (23).
آنیوپلوئیدیهای کروموزومی (Chromosomal Aneuploidies): آنیوپلوئیدیهای کروموزومی از اولین علل ژنتیکی شناختهشدهCHD هستند (24). آنیوپلوئیدیها، تغییراتی در تعداد کروموزومها هستند که معمولاً به صورت de novo (نوظهور- یعنی از قبل در والدین وجود ندارند) و از طریق ناهنجاری در تقسیمات میوزی یا میتوزی ایجاد میشوند. اصلیترین تریزومیهای اتوزومی موجود در جمعیت انسانی، یعنی تریزومی کروموزومهای ۱۳، ۱۸ و ۲۱، با CHD مرتبط هستند، همانطور که سندرم ترنر (مونوزومی کروموزوم X- Turner syndrome) نیز چنین است. در جدول ۲، درصد بیماران دارای فنوتیپ CHD و همچنین انواع CHD دیده شده در چهار سندرم آنیوپلوئیدی مورد اشاره آورده شده است. سندرمهای ناشی از انواع آنیوپلوئیدیها، نفوذپذیری کاملی دارند، اما شدت بیان CHD در آنها متغیر است. به عنوان مثال، تنها در ۴۰ الی ۵۰ درصد افراد با تریزومی ۲۱ (کامل یا جزئی)، CHD از نوع نقص دیواره دهلیزی-بطنی (Atrioventricular Septal Defect) و در بخشی از مبتلایان به سندرم ترنر، تنگی آئورت (Coarctation of the Aorta) مشاهده میشود. با وجود اینکه علت ژنتیکی CHD مرتبط با آنیوپلوئیدیها شناخته شده است، مکانیسمهای مولکولی که باعث اختلال در تکوین قلب میشوند و منجر به نفوذپذیری متغیر CHD میشوند، هنوز بهطور کامل توضیح داده نشدهاند. تحقیقات جدید برای درک این مکانیسمها، بر دستورزی ژنها و مهندسی ژنتیک متمرکز هستند. آنیوپلوئیدیها به ندرت در کشت سلولی مورد مطالعه قرار میگیرند، زیرا سلول واجد آنها در کشت سلولی زنده نمیماند و این موضوع باعث میشود بیشتر پژوهشهای مرتبط با CHD در آنیوپلوئیدیها به مدلهای جانوری و مدلهای انسانی (مانند ارگانوئیدها) متکی باشند (26،27). سازمان ملی ثبت بیماریهای قلبی مادرزادی آلمان (kompetenznetz-ahf.de) که بزرگترین دفتر ثبت CHD در اروپا است، با استفاده از کد بینالمللی IPCCC (International Pediatric and Congenital Cardiac Code) برای دستهبندی انواع CHD و همچنین آنالیز بیماران سندرم داون (Down syndrome) نشان داده است که شیوع انواع CHDها از قبیل تترالوژی فالو، نقص دیوارهی دهلیزی-بطنی، نقص دیوارهی بین دهلیزی و نقص دیوارهی بین بطنی، در افراد مبتلا به تریزومی 21 همواره بالا بوده است (28،29). علاوه بر مطالعات ثبت (Register Study)، روش دیگر برای توصیف ارتباط ژنوتیپ-فنوتیپ در آنیوپلوئیدیها؛ بررسی یک فنوتیپ خاص و سپس توصیف ارتباطات آن با دلایل و علل ژنتیکی است. برای مثال، یک گروه تحقیقاتی، از طریق بررسی گسترده متون علمی نشان دادند که بیماری خروجی دوگانه از بطن راست یا Double outlet right ventricle (DORV) بهطور معمول با تریزومیهای 13 و 18 مرتبط است (30). به دلیل آنکه آنیوپلوئیدیها بهطور معمول در دوران بارداری مورد غربالگری قرار میگیرند، زنان باردار بالای 35 سال، به دلیل ارتباط شیوع بالاتر آنیوپلوئیدیها با افزایش سن مادر، همگی تحت اکوی قلبی جنینی نیز قرار میگیرند. این بررسی میتواند از طریق آمنیوسنتز، تست غیرتهاجمی پیش از تولد یا Non-invasive prenatal testing (NIPT)، کاریوتایپ، آنالیز میکروآرایه (Microarray analysis)، و توالییابی نسل جدید یا Next-Generation Sequencing (NGS) انجام شود (31).
شکل ۱. جدیدترین آمار میزان مرگ و میر نوزادان ناشی از بیماریهای مادرزادی قلبی طبق گزارش GBD سال ۲۰۲۰.
کشور ایران، از مناطق با میزان SDI متوسط تا بالا، جزو مناطق با بالاترین امار مرگو میر ناشی ازین بیماری (رنگ قرمز) نمایش داده شده است (21).
جدول۱: توصیف و دستهبندی شایعترین بیماریهای مادرزادی قلب در کودکان
جدول۲: سندرمهای آنیوپلوئیدی که فنوتیپ CHD در مبتلایان آنها دیده میشود (25)
اختصارات؛ VSD: نقص دیواره بین بطنی، TOF: تترالوژی فالو، AVSD: نقص دیواره دهلیزی-بطنی، CoA: تنگی آئورت، ASD: نقص دیواره بین دهلیزی، DORV: خروجی دوگانه از بطن راست، BAV: دریچه آئورت دو لتی، HLHS: سندروم هیپوپلازی قلب چپ، AS: تنگی دریچه آئورت، TGA: جابجایی عروق بزرگ، PS: تنگی دریچه ریوی.
تغییرات تعداد نسخه CNV: واریانتهای تعداد نسخه یا Copy Number Variation (CNV) از جمله ناهنجاریهای ساختاری کروموزومها هستند و حداقل 10-12 درصد از موارد بیماری قلبی مادرزادی را شامل میشوند (32). سندرمهای ناشی از CNVهای با علائمCHD میتوانند به صورت de novo یا ارثی ایجاد شوند و در جمعیت عمومی بسیار نادر هستند. تاکنون، تغییرات ساختاری مرتبط با CHD در مطالعات خانوادگی و گروهی (cohort) مورد بررسی قرار گرفتهاند. سندرمهای CNV ارثی، ممکن است در والدین ناقل بدون علامت باشند و در جمعیت عمومی مشاهده شوند، بنابراین احتمالاً عوامل ژنتیکی، اپیژنتیکی و محیطی وجود دارند که منجر به نفوذپذیری متغیر بیماری میشوند (29). یکی از سندرمهای CNVمهم، حذف ناحیهی کروموزومی 22q11.2 است که باعث سندرم ولوکاردیوفاشیال (Velocardiofacial syndrome) یا سندرم دیجورج (DiGeorge syndrome) میشود. حذف کروموزومی در ناحیه 22q11.2، دومین علت شایع CHDسندرمی پس از تریزومی 21 است و به تنهایی حدود 4 درصد از موارد CHD را شامل میشود. با این حال، همه افراد دارای سندرم حذف در ناحیه 22q11.2، علائم CHD را ندارند (معمولاً تنها 60-70 درصد موارد دارای علایم بیماری هستند) که نشاندهندهی نیاز به تحقیقات بیشتر در مورد اثر تعدیلکنندههای ژنتیکی و غیرژنتیکی (genetic and non-genetic modifiers) بر نفوذ بیماری CHD است (33). مطالعات مورد-شاهدی CHD خانوادگی میتوانند CNVهای ارثی مرتبط با خطر CHD را شناسایی کنند. برای مثال، در یک خانواده با تکرار موارد بیماری تترالوژی فالو بین اعضا آن خانواده، حذف 1/8 مگابازی در موقعیت کروموزومی 10p11 شناسایی شد. این موقعیت کروموزومی دربرگیرنده ژن مهمی است که گیرنده همکار فاکتور رشد اندوتلیال عروقی یا Vascular endothelial growth factor (VEGF) co-receptor به نام نوروپیلین ۱ یا Neuropilin-1 (NRP1) را کد میکند. در سلولهای آمنیوسیت، یکی از اعضای این خانوادهی مبتلا، سطح کاهشیافته بیان ژن NRP1 نیز مشاهده شد (34،35). علاوه براین، با مقایسهی مناطق حذف کروموزومی در افراد مبتلا به سندرم حذف 10q26، ژن WDR11 به عنوان یک ژن کاندید جدید برای سه فنوتیپ بیماری CHD، بیماری کلوبوما (Coloboma، نقص مادرزادی ساختار چشم که به دلیل بسته نشدن کامل شکاف جنینی در دوران رشد ایجاد میشود) و تأخیر رشدی جنین شناسایی شد. کشف واریانتهای تک نوکلئوتیدی در این ژن که همپوشانی فنوتیپی با سندرمهای حذفی در همان ژن را دارند، همانند نقش تائید شده ژن MEIS2، میتواند نقش آن ژن در بروز CHD را اثبات کند. پروتئین کدشده توسط این ژن به نام Meis Homeobox 2 متعلق به خانواده homeobox میباشد که در تنظیم بیان ژنهای مهمی در طی تکوین رویانی نقش دارد. چنین مطالعاتی، مناطق ژنومی کاندید را شناسایی میکنند که ممکن است ردههای سلولی تکوینی متعددی را تحت تاثیر قرار دهند (34). همچنین مشخص شده است که حذفهای هموزیگوت در ژن PCDHA که کدکننده پروتئین پروتوکادهرین آلفا (Protocadherin α) میباشد با بیماری انسداد مجرای خروجی بطن چپ یا Left ventricular outflow tract obstruction (LVOTO) مرتبط است، به طوری که این تغییرات ساختاری کروموزومی، در نیمی از گروه مورد مطالعه مبتلا به تنگی آئورت ایزوله و در ۱۶ درصد از افراد گروه کنترل مشاهده شد. در بررسی آرایههای پلیمورفیسم تکنوکلئوتیدی (single nucleotide polymorphism arrays) در 2539 فرد مبتلا به CHD و 1538 فرد بدون CHD، در مبتلایان افزایش 1/8 برابری در حذفهای نادر نواحی کدکننده مشاهده شد. همچنین در ناحیه کروموزومی 15q11.2 حذفهایی در ژنهای مرتبط با مسیر پیامرسانی Wnt گزارش شد (38-36). بنابراین، تغییرات ساختاری که بهطور حتم با CHD مرتبط هستند، شامل حذف یا تکثیر در ناحیه کروموزومی 1q21.1، حذف 1p36، حذف یا تکثیر 7q11.23، حذف یا تکثیر 8p23، حذف 10q، حذف 11qter حذف 15q11.2، حذف 15q25.2، حذف یا تکثیر 16p11.2 و حذف یا تکثیر22q11 هستند (41-39). جدول ۳ سندرمهای مرتبط با تغییرات تعداد نسخه، درصد بیماران دارای علائم CHD و ژن یا ناحیه کروموزومی دچار تغییر را نمایش میدهد. واریانتهای تک ژنی یا Single Gene Variants (SGVs): این گروه از واریانتها در ژنهای مرتبط با سندرمهای تکژنی از جمله؛ فاکتورهای رونویسی، عوامل پیامرسان داخل سلولی، مولکولهای چسبندگی سلولی و پروتئینهای ساختاری قلب رخ میدهند (43-25). بیماریهای قلبی مادرزادی که ناشی از نقصهای ژنی تکگانه با الگوهای وراثتی مندلی هستند و اغلب بهصورت سندرمی ظاهر میشوند، سهم کوچکی از انواع بیماری CHD در اثر عوامل ژنتیکی را تشکیل میدهند. واریانتهای نوظهور، حدود ۸ درصد از کل موارد CHD را شامل میشوند، در حالیکه تغییرات تکنوکلئوتیدی اتوزومال مغلوب یا single-nucleotide variants (SNVs) و جهشهای درجی- حذفی (indels) تنها حدود ۲ درصد از این موارد را در بر میگیرند (15). اگرچه بسیاری از علل مونوژنیک CHD هنوز شناسایی نشدهاند، مطالعه ژنهای شناختهشده مرتبط با CHD و محصولات پروتئینی آنها، بینشهای ارزشمندی را درباره تکوین و زیستشناسی قلب فراهم کرده است. نکتهی مهم این است که بیشتر توالییابیهای ژنوم انسان در نمونههای خون محیطی بیماران انجام میشود که این روش ممکن است تغییرات موزائیک یا سوماتیک موجود در بافتهای قلبی را شناسایی نکند (44). مطالعات ژنتیکی انسانی با استفاده از تحلیلهای کشف ژنهای جدید و مرتبط، درک ما را از واریانتهای نادر در نواحی کدکنندهی پروتئین مرتبط با CHD بهبود بخشیدهاند. جدول ۴ سندرمهای مرتبط با CHD که در اثر جهشهای تکژنی نقطهای بهوجود آمدهاند، ژن دچار جهش در آن سندرم و درصد بیماران دارای فنوتیپ CHD را نمایش میدهد. این تحلیلها شامل مطالعات همراهی سراسر ژنوم یا بررسی فهرستهای ویژهای از ژنهای کاندید هستند (29). به عنوان مثال، واریانتهای نادر متعدد در ژنهای NOTCH1 و FLT4 در افراد مبتلا به تترالوژی فالو یافت شده است (14). افزایش استعداد ابتلا به CHD با واریانتهای نادر در ژنهای مرتبط با سندرمهای ژنتیکی و فنوتیپهای دیگر غیر از فنوتیپ نقایص قلبی مانند ژن POLR1A (در اختلالات جمجه-صورت)، ژن KDM2B (در اختلالات تکوین سیستم عصبی) و ژن PPP2R1A (در اختلالات تکوین سیستم عصبی) شناسایی شده است. همچنین، با توجه به همزمانی قابل توجه بروز CHD و اختلالات تکوین سیستم عصبی، واریانت ژن TKT، کدکنندهی ترنس کتولاز (Transketolase) در افراد خانوادهای با ویژگیهای تأخیر رشدی، کوتاهقدی و CHD شناسایی شد (45). وجود واریانت در گروههای مبتلا به نوروفیبروماتوز نوع ۱ یا سندرم Ellis-Van Creveld نیز ارتباطات خاص ژنوتیپ-فنوتیپ با ناهمگنی فنوتیپی یک سندرم را تائید مینماید (46). برخی از مطالعات بر روی ژنهای خاص شناساییشده در بیماریهای قلبی مادرزادی خانوادگی تمرکز کرده و از تفاوتهای فنوتیپی اعضا برای بررسی اساس چندژنی این بیماری استفاده میکنند. بهعنوان مثال، یک حذف نادر کوچک در ژن TPM1، که کدکننده زنجیرهی آلفا-تروپومیوزین (α-tropomyosin) است، در یک خانواده با نقص دیواره دهلیزی که در پنج نسل متوالی مشاهده شده بود، از طریق آنالیز پیوستگی (Linkage Analysis) در سطح ژنوم شناسایی شد. قبل انجام این آنالیز، با استفاده از توالییابی کل ژنوم واریانتهای خاص پیدا شده بود. کاهش بیان Tpm1 در طول تکوین، از طریق تکنیک مورفولینو (Morpholino antisense oligonucleotide) و کریسپر یا Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) در مدل Xenopus tropicalis، منجر به ادم قلبی و نقص دیوارهی دهلیزی شد. حذف ژنی Tpm1کدکننده زنجیرهی آلفا-تروپومیوزین (α-tropomyosin) با روشCRISPR-Cas9 در موشها، با خمیدن غیرطبیعی لولهی قلبی اولیه و مرگ زودهنگام رویان همراه بود، که ضرورت وجود آلفا-تروپومیوزین در تکوین قلب مهرهداران را تائید میکند (47). ژن NOTCH1 نیز که گیرنده پروتئین Notch (Notch Receptor 1) را کد میکند، مثال دیگری است که واریانتهای آن طی تکنیک توالییابی اگزوم به روش paired-end در مبتلایان به سندرم قلب چپ هیپوتروفیک که سایر ناهنجاریهای مادرزادی را نداشتند، شناسایی شد (48). واریانتهای نادر مغلوب، مانند جهشهای دوگانه در NKX2-6 (کد کننده پروتئین هومئوباکس Nkx-2.6 یا Homeobox protein Nkx-2.6) و TMEM94 (کد کننده پروتئین تراغشایی ۹۴ یا Transmembrane protein 94) نیز بهعنوان علل ایجاد CHD شناسایی شدهاند (49). نقص در پروتئین TMEM94 به عنوان یک علت شناختهشده برای اختلالات تکوینی سیستم عصبی شناخته میشود (49)، اما در اعضای چند خانواده دارای واریانت این ژن، فنوتیپ بیماری مادرزادی قلب هم وجود داشت. روش توالییابی اگزوم نیز دو واریانت متمایز در این ژن را در هر افراد مبتلا به CHD شناسایی کرد که توارث دوآللی داشتند. همچنین، توالییابی RNA از رده سلولی لنفوبلاستوئید (Lymphoblastoid cell lines) از اعضای یک خانوادهی مبتلا، تأثیر واریانتهای محل پیرایش (splice site variants) بر بروز CHD را تأیید کرد (49). علاوه بر مطالعات ژنومی روی گروههای انسانی، سلولهای بنیادی پرتوان القاشده یا Induced Pluripotent Stem Cells (iPSC) میتوانند بهعنوان مدل in vitro برای بررسی تکوین قلب استفاده شوند. بهعنوان مثال، واریانت بدمعنی p.R443P در پروتئین MYH6 از زیرواحدهای پروتئین میوزین (Myosin)، که به عنوان واریانت بیماریزا و مرتبط با سندرم قلب چپ هیپوتروفیک شناسایی شده بود، در یک تحقیق با استفاده از CRISPR-Cas9 و نوترکیبی همولوگ در iPSCهای انسانی ایجاد شد تا تمایز این سلولها به کاردیومیوسیت یا سلول ماهیچه قلبی (cardiomyocyte) تحت اثر این واریانت مورد مطالعه قرار بگیرد. این سلولهای بنیادی پرتوان القایی به کاردیومیوسیتها تبدیل شدند و با استفاده از CHIR99021 و Activin-A (دو ترکیب شیمیایی مورد استفاده برای حفظ ویژگی بنیادی سلول) و در محیط RPMI/B27 بدون انسولین کشت داده شدند. پس از اصلاح واریانت p.R443P در پروتئین MYH6، بهبود تمایز کاردیومیوسیتها و ساختار سارکومر در رده سلولی فرد پروباند مشاهده شد. این مطالعه بر اهمیت سلولهای بنیادی پرتوان القایی مشتق از بیماران و ویرایش ژنومی آنها در ارزیابی عملکرد بیماریزایی واریانتهای پرخطر بیماری قلبی مادرزادی تأکید میکند (29). جهشهایی در ژنوم میتوکندری در مبتلایان به بیماریهای مادرزادی قلبی نیز شناسایی شدهاند که ممکن است در بیماریزایی این اختلالات قلبی دخیل باشند (50،51).
فاکتورهای رونویسی اختصاصی قلب (Cardiac Transcription Factors): تشکیل قلب یک فرآیند پیچیده و نیازمند تعامل بین انواع سلولهای متمایز و وابسته، از طریق مسیرهای پیامرسانی و مسیرهای رونویسی در مکان و زمان خاص است که به تمایز و تعیین سرنوشت آنسلولها منجر میشود (شکل ۲-الف و ۲-ب). اختلال در این فرآیند تکوینی میتواند باعث بروز بیماریهای مادرزادی قلب شود. برنامه تنظیم بیان ژنهای اختصاصی تکوین قلب، توسط فاکتورهای رونویسی که بیان ژنهای کدکننده فاکتورهای رونویسی دیگر را تنظیم کرده و شبکههای خاصی مانند GATA4، NKX2-5 و TBX5 را تشکیل میدهند، هدایت میشود. جهشهای بیماریزا در هر یک از فاکتورهای رونویسی مهم در تکوین رویانی قلب، موجب بر همخوردن این برنامهی بسیار دقیق و پیچیده و ایجاد انواع نقصهای ساختاری قلب خواهد شد (43، 55-52). جدول ۵، بیست مورد از فاکتورهای رونویسی با بالاترین امتیاز «ارتباط ژن-بیماری یا Gene-Disease Interaction (gdi)» را به همراه تعداد واریانتهای بیماریزای آنها نمایش داده است. اختلال در دو فاکتور رونویسی اختصاصی و ضروری تکوین قلب، به نامهای GATA4 و TBX5 از اولین علل ژنتیکی شناختهشدهی CHD خانوادگی میباشند. واریانتهای بیماریزای هتروزیگوت در TBX5 باعث نقص در جدایی حفرهها و تشکیل دیواره و سایر اشکال CHD در سندرم هولت-اورام (Holt-Oram) میشوند. همچنین، واریانتهای هتروزیگوت در ژن GATA4 باعث نقصهای دیواره دهلیزی- بطنی، تنگی شریان ریوی، و ناهنجاری مجراهای خروجی قلب میشوند. اختلال در برهمکنش فیزیکی بین این دو پروتئین یا برهمکنش با سایر کوفاکتورهای اختصاصی در اثر رخداد واریانتهای بدمعنی در آنها، باعث ناهنجاریهای شدید قلبی میشود (56).
جدول۳ : سندرمهای ناشی از ریزحذف و تغییرات تعداد کپی یا CNV همراه با فنوتیپ CHD (9،22،49).
جدول۴: سندرمهای مرتبط با CHD که در اثر جهشهای تک ژنی نقطهای بهوجود آمدهاند (9،22،42).
شکل ۲: تکوین قلب انسان (الف) نمایش شماتیک مراحل تکوین انسان و نقاط عطف کلیدی آن. سلولهای ناحیه اولیه قلب FHF(آبی) و ناحیه ثانویه قلب SHF(نارنجی) سلولهای تاج عصبی قلبی CNCCs(سبز) و اندام پرو اپیکاردیال PEO (بنفش) و بخشهای مشتق از آنها به تصویر کشیده شدهاند. در هفته دوم، مزودرم قلبی به پیشسازهای FHF و SHF تبدیل میشود که ساختار هلال قلب را تشکیل میدهند. FHF لوله اولیه قلب (PHT) را تولید میکند که به ترتیب به بطن چپ (LV) و قسمتهایی از دهلیزهای راست و چپ تبدیل میشود. سلولهای SHF در رشد بطن راست (RV)، مجرای خروجی (OFT)، دهلیزها و میوکارد ورودی نقش دارند. سلولهای PEO در تشکیل اپیکارد و رگهای کرونری مشارکت دارند. سلولهای CNCCs از لوله عصبی پشتی به OFT قلب مهاجرت میکنند و در تشکیل دیواره، جدایی شریانهای آئورت و ریوی و تشکیل دریچهها و توزیع عصب پاراسمپاتیک نقش دارند. (ب) مروری بر مسیرهای اصلی پیامرسانی و مهمترین عوامل رونویسی که هر مرحله ذکر شده از تکوین قلب را تنظیم میکنند. مسیر غیرکلاسیک WNT با ncWNT نشان داده شده است (8).
جدول۵: فاکتورهای رونویسی قلبی مهم که به ترتیب بیشترین امتیاز ارتباط ژن- بیماری مادرزادی قلبی (gdi) را دارا هستند. تعداد بسیاری از دیگر فاکتورهای رونویسی دخیل در CHD در این جدول نمایش داده نشدهاند.
مسیرهای پیامرسانی داخل سلولی: شبکهای بسیار دقیق و هوشمند، شامل مسیرهای پیامرسانی Nodal، Transforming Growth Factor-β (TGFβ)، Bone Morphogenetic Protein (BMP)، WNT و رتینوئیک اسید (Retinoic acid)، فرآیند فعالسازی بیان ژنهای مرتبط با فاکتورهای رونویسی قلبی را هدایت میکنند. این فاکتورها، از جمله NKX2-5، GATA4/5/6، و TBX1/5/20، برای تبدیل سلولهای پیشساز نواحی اولیه و ثانویه قلب به سلولهای تخصصی مرتبط با حفرههای مختلف قلب ضروری میباشند (57). مسیر پیامرسانی Notch در فرآیند تکوین دریچههای قلب و حفظ هموستازی آنها نقش حیاتی ایفا میکنند (58). فاکتور پروتئینی رشد اندوتلیال عروقی یا Vascular endothelial growth factor A (VEGF-A) به عنوان یک میتوژن کلیدی، در تشکیل دیواره دریچههای دهلیزی- بطنی، مانند دریچههای تریکاسپید (tricuspid valve) و میترال (mitral valve) نقش دارد. تغییر در سطح بیان این پروتئین در مراحل تکوین قلب، چه افزایشی باشد و چه کاهشی، با نقصهای مادرزادی قلبی مانند نقص دیواره دهلیزی- بطنی مرتبط است (59). همچنین، در مسیر پیامرسانی Hedgehog بیان ژن Sonic hedgehog (SHH) برای تشکیل دیوارهی بین بطنی قلب ضروری است. در مراحل اولیه تکوین رویان، این مسیر باعث مهاجرت سلولهای ستیغ عصبی قلبی به مکان اولیه تشکیل قلب، یعنی مجرای خروجی قلب (cardiac outflow tract) میشود، بگونهای که سلولهایی که سیگنال SHH را دریافت میکنند، فاکتور رونویسی قلبی GATA4 را در ناحیه ثانویه قلبی یا second heart field (SHF) فعال میسازند (60). پروتئین BMP که در مسیر ابرخانواده سیتوکاینهای TGF-β عمل میکند، از طریق فعالسازی فاکتور رونویسی Smad، نقش ضروری در تشکیل صفحه اولیه قلبی یا first heart field (FHF) ایفا میکند (61). رتینوئیک اسید نیز با محدودکردن رشد در ناحیهی ثانویه قلبی، در تنظیم تکوین قلب مشارکت دارد. جهش در ژن Raldh2، که مسئول سنتز رتینوئیک اسید است، این محدودیت رشدی را از بین میبرد، در نتیجه، بخشهایی از قلب که از ناحیهی ثانویه قلبی منشأ میگیرند، با نقصهای ساختاری مواجه خواهند شد (64-62). شکل ۲-ب مسیر پیامرسانی دارای نقش عملکردی مهم در هر مرحله از تکوین قلب رویان را نمایش میدهد.
نقش عوامل اپیژنتیک (Epigenetics) در تکوین قلب: پژوهشهای متعددی نشان دادهاند که تغییرات اپیژنتیک (epigenetic modification) از طریق مکانیسمهایی مانند متیلاسیون DNA (DNAmethylation)، نوسازی یا رمدلینگ ساختار کروماتین (Chromatin remodeling)، تغییرات هیستونی (Histone modifications) و تنظیم بیان ژنها (gene expression regulation) توسط RNAهای غیرکدکنندهی کوچک (MicroRNA) و بزرگ Long-noncoding RNAs))، بدون تغییر در توالی نوکلئوتیدی DNA، بیان ژنهای دخیل در تکوین قلب را کنترل میکنند و نقشی کلیدی در ایجاد CHD دارند (70-65، 57). متیلاسیونDNA (DNA methylation): متیلاسیون DNA، یکی از نخستین مکانیسمهای کشفشده اپیژنتیکی است که شامل افزودن گروه متیل به نوکلئوتیدهای سیتوزین در جزایر CpG (CpG islands) است و در فرایندهای بیولوژیکی مختلف از جمله تکوین قلب نقش دارد. در طی رشد رویان، بلوغ کاردیومیوسیتها با تغییرات متیلاسیون DNA همراه است. ابتدا، یک موج دمتیلاسیون (demethylation) در ژنهای قلبی، بهویژه ژنهایی که پروتئینهای سارکومر (sarcomere) را کد میکنند، رخ میدهد و پس از تولد نوزاد، روند متیلاسیون مجدد، شکل میگیرد. هرگونه تغییر غیرطبیعی در متیلاسیون DNA میتواند از طریق تأثیر بر بیان ژن، به بروز CHD منجر شود (71). در این فرآیند، فولات (Folate) یا ویتامین B9 بهعنوان منبع اصلی گروههای متیل برای متیلاسیون DNA، نقشی حیاتی دارد. مطالعات نشان دادهاند که مصرف مکمل فولات توسط مادران میتواند خطر ایجاد CHD را در نوزادان مبتلا به سندرم داون (Down syndrome) کاهش دهد. آنزیم متیلنتتراهیدروفولات ردوکتاز یا Methylenetetrahydrofolate reductase (MTHFR) که در متابولیسم فولات نقش کلیدی دارد، در صورت کاهش فعالیت میتواند منجر به ایجاد شکستگی در DNA و تفرق غیرطبیعی کروموزومها شود. پژوهشها نشان دادهاند که افزایش متیلاسیون پروموتر MTHFR در مادران دارای فرزندان مبتلا به CHD و سندرم داون بهطور قابلتوجهی افزایش یافته است. همچنین، در کودکان مبتلا به CHD، سطح بالاتری از نشانگرهای زیستی متیلاسیون مشاهده شده است که این افزایش با ناهنجاریهای پیچیدهی قلبی در ارتباط است (72).
نوسازی ساختار کروماتین (Chromatin remodeling): نوسازی (رمدلینگ) ساختار کروماتین یکی دیگر از مکانیسمهای مهم اپیژنتیک است که وابسته به فعالیت چهار کمپلکس مرتبط با ATP میباشد: کمپلکس SWI/SNF (SWItch/Sucrose Non-Fermentable)، پروتئین Chromodomain helicase DNA-binding (CHD) ، پروتئین Iswi و پروتئینINO80 . حذف پروتئین BRG1 (یک فعالکننده رونویسی کد شده توسط ژن SMARCA4)، یکی از اجزای کمپلکس BRG-/BRM-associated factor (BAF) که همولوگ کمپلکس SWI/SNF در مهرهداران است، موجب نقص در تشکیل دیواره بین بطنی و همچنین ناهنجاریهایی در بطن راست و مجرای خروجی قلب میشود. کاهش مقدار Baf60c، یکی دیگر از اجزای کمپلکس BAF، در موشها منجر به نقص در نواحی قدامی قلب، اختلال در خمیدگی لوله قلبی در طی تکوین جنین، کوتاهی مجرای خروجی قلب و هیپوپلازی بطن میشود. علاوه بر این، ناکفایتی هاپلوئیدی ژن CHD7 با سندرم CHARGE مرتبط است که در مبتلایان آن، نقصهای دیواره بین دهلیزی، دیوارهی بین بطنی، دیواره دهلیزی-بطنی و ناهنجاریهای کونوتراکال قلب (conotruncal heart defects) دیده میشود (73,74).
تغییرات شیمیایی هیستونها (Histone modifications): مکانیسمهای تغییرات هیستونی شامل اثر هیستونداستیلازها یا Histone deacetylases (HDACs)، هیستوناستیلازها یا Histone acetyltransferases (HATs) و هیستونمتیلترنسفرازها یاHistone methyltransferases (HMT) بر DNA میشوند. حذف پروتئینهای HDAC5 و HDAC9 در سلولهای رده زایا با نقص دیواره بین بطنی مرتبط است (75). حذف SMYD1 که یک متیل ترانسفراز هیستونی است موجب هیپوپلازی بطن میشود. حذف WHSC1، یک متیل ترانسفراز هیستونی دیگر، با نقصهای دیواره بین دهلیزی و دیواره بین بطنی مرتبط است. همچنین، حذف هیستوندمتیلاز در سلولهای رده زایا با ناهنجاریهای پیچیدهای، مانند خروجی دوگانه بطن راست و هایپرتربکولاسیون (hyper trabeculation) همراه است (76). نواحی غیر کدکننده (Non-coding) ژنوم: تنها 1 تا 2 درصد از DNA ژنومی انسان از اصل «Central dogma» پیروی میکند، به این معنی که DNA به RNA رونویسی میشود و سپس به پروتئین ترجمه میشود. حدود ۹۸ درصد دیگر، کدکننده پروتئین نیستند که به آن DNA غیرکدکننده (Non-coding DNA) نیز گفته میشود. حدود ۲۰ درصد از این DNA که پیش از این از نظر عملکردی «بیفایده یا junk» خوانده میشدند، در میان مهرهداران مختلف بسیار حفاظت شدهاند و حدود 80 درصد آنها عملکردهای بیوشیمیایی مهمی در سلول دارند (77). RNAهای غیرکدکننده در سالهای اخیر بهعنوان عوامل کلیدی در افزایش خطر ابتلا به انواع بیماریهای انسانی، از جمله بیماریهای مادرزادی قلب، شناسایی شدهاند. این مولکولها با تنظیم فاکتورهای رونویسی، تکثیر کاردیومیوسیتها، و همچنین تمایز و تکوین عضلات قلبی و اسکلتی، نقش مهمی در فرآیندهای زیستی ایفا میکنند (81-78). بخش عظیمی از تحقیقات کنونی، در حال تلاش برای شناسایی عملکردهای بیوشیمیایی جدید برای DNAهای غیرکدکننده است، تا دادههای حاصل از تحقیقات را به فنوتیپهای مولکولی، فیزیولوژیکی و پاتولوژیکی مرتبط کند. تا پیش از این، به دلیل کمبود دادههای ژنوم انسان و عدم وجود الگوریتمها و روشهایی برای ارتباط DNA نادر یا نوظهور با عملکرد و فنوتیپ بیماران، بیماریزایی واریانتهای غیرکدکننده قابل بررسی نبود. با اینحال، برخی از مطالعات، شواهدی ارائه میدهند که بخشهای غیرکدکننده ژنوم، برای تکوین قلبی-عروقی ضروری هستند (82). بهعنوان مثال، واریانتهای نادر هموزیگوت غیرکدکننده در سندرم هولت-اورام، با ایجاد اختلال در عناصر تنظیمکننده اختصاصی قلب، باعث بروز CHDهای ایزوله میشوند (83). همچنین شواهدی وجود دارد که نشان میدهند واریانتهای نادر غیرکدکننده نوظهور در نزدیکی ژنهای شناختهشده مرتبط با CHD، در گروههای بیماران، نسبت به گروه کنترل، بیشتر مشاهده میشوند و حتی زیرمجموعهای از این واریانتهای غیرکدکننده نوظهور، بیان ژنهای CHD را نیز تغییر میدهند )84(. اگرچه مطالعات همراهی در سطح ژنوم، از قدرت آماری محدودی برخوردارند، اما تاکنون، واریانتهای غیرکدکنندهی شایع در ارتباط با بیماریهای قلبی مادرزادی، بهویژه در نقصهای دیوارهای و نقصهای انسدادی مجرای خروجی بطن چپ شناسایی شدهاند (87-85). اخیراً، یک متا- آنالیز که چهار گروه CHD با 55,342 شرکتکننده را با پنل مرجع TOPMed مقایسه کرده است، 16 موقعیت کروموزومی مرتبط با خطر CHD را شناسایی کرد (88(. از این 16 موقعیت، 13 مورد به ژنهای مرتبط با تکوین قلبی مربوط میشوند. این یافتهها بر اهمیت بیشتر بررسی نقش عملکردی ژنوم غیرکدکننده و درک بهتر ارتباط آن با استعداد ابتلا به CHD تأکید میکنند (89). MicroRNAها (miRNA)و نقش آنها در بروز CHDMicroRNAها: مولکولهای کوچک RNA غیرکدکنندهای هستند (با طول ۱۹ تا ۲۲ نوکلئوتید) که بیان ژن در یوکاریوتها را در سطح پس از رونویسی تنظیم میکنند. این مولکولها در تعامل با ناحیه 3'UTR از RNA پیامرسان (mRNA) ژن هدف، مانع از بیان آن ژن میشوند (91, 90). تولید میکروRNAها در انسان، با رونویسی یک میکروRNA اولیه (pri-miRNA) توسط RNA پلیمراز II آغاز میشود که حاوی ساختاری سنجاقمانند است. این ساختار توسط کمپلکس DROSHA-DGCR8 به یک پیشساز میکروRNA (pri-miRNA) ۶۰ تا 90 نوکلئوتیدی با انتهای چسبنده در 3'UTR پردازش میشود. این پیشساز میکروRNA از طریق پروتئین XPO5 در فرایندی وابسته به RAN-GTP به سیتوپلاسم منتقل میشود، جاییکه توسط DICER (RNase III endoribonucleases) و Transactivation response RNA binding protein (TRBP) به یک دورشتهای 19 تا 22 نوکلئوتیدی تبدیل میشود. نهایتا یک رشته، بر اساس محتوای پورین و پایداری ترمودینامیکی انتهای 5'UTR، انتخاب شده و در کمپلکس RNA-induced silencing complex (RISC) ادغام میشود تا تخریب، ناپایداری یا مهار ترجمه mRNA هدف را هدایت کند (92). برهمکنش miRNA و هدف آن توسط ناحیهی seed region ( شامل نوکلئوتیدهای ۲ تا ۸ در 5'UTR میکروRNA) میانجیگری میشود. محلهای اتصال مکمل و معکوس میکروRNA (Reverse-complementary miRNA binding sites) که بهعنوان عناصر پاسخدهنده به میکروRNA (MREs) نیز شناخته میشوند، معمولاً در نواحی 3'UTR از RNAهای پیامرسان (mRNA) هدف قرار دارند، اما میتوانند در نواحی 5'UTR یا توالی کدکننده پروتئین نیز وجود داشته باشند (92). برخی miRNAها در بین گونههای متنوع جانوری حفاظت شدهاند و در بافتهای خاصی بیان میشوند. این بیان اختصاصی بافتی miRNAها، در رشد، تکثیر و تمایز بافتها، تصمیمگیری برای سرنوشت ردههای سلولی و همچنین homeostasis بافت، اثرگذار است. از میان miRNAهایی که به طور خاص در بافت قلب بیان میشوند، miR-1 به عنوان فراوانترین miRNA در قلب جوندگان شناسایی شده است (حدود 45% از کلmiRNAهای قلبی را شامل میشود). miR-1b، miR-1d، miR-133، miR-206، miR-143 و miR-208، شش miRNA با بیان اختصاصی در عضله اسکلتی و قلب هستند که از بررسی بیان ۱۱۹ میکروRNA در اندامهای انسان و موش یافت شدند (94, 93). در این میان، نقش miR-1، miR-133، و miR-208b در تکوین رویانی قلب انسان تائید شده است (97-95). این miRNAهای اختصاصی عضله، به عنوان MyomiRs نامگذاری شدند و نقش اساسی در تکوین قلب ایفا میکنند (98). برخی از این miRNAها در شکل ۳ در کنار هر مرحلهی تکوینی نمایش داده شدهاند. هرگونه اختلال و جهش جدید در محل اتصال RNAهای غیر کدکننده روی DNA که در نقش و عملکرد تنظیمی آنها تغییری ایجاد کند، میتواند باعث افزایش استعداد ابتلا به CHD شود. بنابراین مطالعۀ نواحی نظیر 3'UTR ژنهای هدف، میتواند یکی از راههای مهم تشخیص مکانیسم پاتوژنز این بیماری و حائز اهمیت مطالعه بیشتر محققان باشد (99،100).
شکل ۳. نقش miRNAها در تنظیم مراحل مختلف رشد قلب. فلشها بیانگر تنظیم فاکتور رونویسی توسط miRNA مربوطه میباشد.
اختصارات: Ao: آئورت، CPC: سلولهای پیشساز قلبی، IVS: سپتوم بینبطنی، L: چپ، LA: دهلیز چپ، LV: بطن چپ، ML: خط میانی، OFT: مسیر خروجی، P: خلفی، FHF: ناحیه اولیه قلبی، PLA: دهلیز چپ اولیه، PRA: دهلیز راست اولیه، PS: خط اولیه، PT: تنه ریوی، R: راست، RA: دهلیز راست، RV: بطن راست، SHF: ناحیه ثانویه قلبی، Tr: ترابکولا (102, 101).
lncRNAها و نقش آنها در بروز CHD: RNAهایی که طولشان بیشتر از 200 نوکلئوتید بوده و به هیچ محصول پروتئینی ترجمه نمیشوند، در یک دسته به نام RNAهای غیر کدکننده بلند Long-noncoding RNAs)) یا lncRNAها گروهبندی میشوند. lncRNAها معمولاً با تنظیمات پیرایشی، برش داده میشوند، یک کلاهک در جهت 5'UTR و دم poly(A) در جهت 3'UTRدارند و الگوهای بیانی وابسته به مکان و زمان در بافتهای متنوع، از جمله بافت قلب از خود نشان میدهند. برخلاف mRNAها، توالی اکثر lncRNAها، بهطور میانگین حفاظتشدگی (conservation) ضعیفی در میان گونههای جانوری دارند که این امر تردیدهای اولیهای را در مورد اهمیت عملکردی آنها، در کنترل فرآیندهای زیستی اساسی، مانند تکوین قلب بهوجود میآورد. در واقع، بهنظر میرسد بیشترlncRNAها، مختص گونه هستند. بهعنوان مثال، بهنظر نمیرسد که تقریباً 70 درصد از lncRNAهای انسانی، همولوگی با توالی مشابه در هیچ گونهی دیگری داشته باشند. از یک سو، این علت میتواند توانایی تحقیق و بررسی عملکردی lncRNAهای خاص انسان را در مدلهای غیرانسانی محدود کند و از سوی دیگر، نشان میدهد که lncRNAهای غیرانسانی، به دلیل عدم حضور توالی مشابه آنها در ژنوم انسان، برای بروز بیماری در انسان ضروری نیستند. با اینحال، بیش از هزار همولوگ lncRNA انسانی احتمالی در سایر گونهها شناسایی شده است که نشان میدهد بسیاری ازlncRNAها ممکن است برای تنظیم فرآیندهای زیستی حفاظتشدهای اهمیت داشته باشند. در واقع، مطالعات in vivo که در سالهای اخیر بر روی lncRNAها انجام شدهاند، هیچ شکی باقی نگذاشتهاند که lncRNAها برای سلامت قلبی- عروقی جنین، از اهمیت زیادی برخوردارند، بهطوری که نقش چندین lncRNA برای تکوین قلب به اندازهی نقش پروتئینهای حیاتی قلبی ضروری است. همانند سایر بافتها، lncRNAها در تمام انواع سلولهای قلب، از جمله کاردیومیوسیتها، سلولهای اندوتلیال، سلولهای عضله صاف عروقی و فیبروبلاستها یافت میشوند. lncRNAها در تمام مراحل تکوین قلب بیان میشوند و بهطور دینامیک و پویایی تنظیم میشوند. lncRNAها بهعنوان کمک-تنظیمکنندههای (co-regulators) شبکههای رونویسی و مسیرهای پیامرسانی، متشکل از پروتئینها و RNAهای غیرکدکننده دیگر، مشارکت دارند و همراه با اجزای دیگر این شبکه تنظیمی، الگوی دقیق بیان ژنهای مورد نیاز برای تکوین طبیعی قلب در موقعیت و زمانهای مشخص را هماهنگ میکنند. هرگونه اختلال در این شبکههای تنظیمی، میتواند تاثیرات ویرانگری بر تکوین اولیهی قلب داشته باشد. اختلال در lncRNAهای قلبی در طول مورفوژنز اولیه قلب، میتواند منجر به نقص در تعهد ردههای سلولی قلبی (cell lineage commitment)، روند تشکیل بطن و جداشدن صحیح دیوارهها در قلب شود (104, 103). بهطور کلی، ایجاد پیشساز miRNA، پایدارکنندگی ساختار miRNA، رقابت با miRNA برای تنظیم رونویسی، جذب miRNA و اتصال به آن و در نتیجه مهار کردن فعالیت تنظیمی miRNA، ایجاد داربست و برهمکنش با پروتئین و DNA و RNA (تشکیل کمپلکس ریبونوکلئوپروتئین)، تشکیل ساختارهای تریپلکس (triplex) با اتصال به دو رشتهای DNA و عمل به عنوان راهنمای جابجایی فاکتورهای تنظیمی در سلول، از جمله نقشهای تنظیمی lncRNAها هستند. همچنین در حال حاضر، بسیاری از lncRNAهای تنظیمکننده تکوین، تکثیر و تمایز بافتی، با منشأ مزودرمی یافت شدهاند که از جهت مطالعۀ روند ایجاد بیماری، به عنوان بیومارکرهای مولکولی قابل توجه هستند (106, 105). در جدول 6، شماری از lncRNAهای اثرگذار در تکوین قلب به همراه نقش آنها نام برده شده است و در شکل ۴ بخشی از عملکردهای سلولی lncRNA به تصویر کشیده شده است (103).
جدول۶: مشخصات برخی از lncRNAهای مرتبط با تکوین قلب با عملکردهای شناسایی شده.
شکل ۴. انواع نقشهای عملکردی lncRNA درسلول. lncRNA در تعامل با DNA، پروتئین، miRNA و mRNA انواع نقشهای تنظیمی را ایفا میکند.
نتیجهگیری
مطالعه حاضر، دیدگاهی جامع در مورد عوامل ژنتیکی و اپیژنتیکی مؤثر بر تکوین قلب و بیماریهای مادرزادی قلب (CHD) ارائه داده است. پیشرفتهای اخیر در ویرایش ژنوم و بهرهگیری از قابلیتهای سلولهای بنیادی پرتوان، امکان شناسایی دقیق عملکرد ژنهای اثرگذار بر تکوین قلب را فراهم کردهاند. با اینکه هنوز تمام جنبههای بیماریزایی ژنهای مرتبط با CHD بهطور کامل شناخته نشده است، روشهای جدید تشخیصی و بهبود طراحی آزمایشها، نویدبخش یافتن مکانیسمهای مولکولی کلیدی در ایجاد این بیماری هستند. این تحقیقات بینشی جامع پیرامون فرآیندهای مرتبط با تکوین قلب در اختیار متخصصان، محققان، پزشکان و سیاستگذاران حوزه سلامت خواهد گذاشت. همچنین، این یافتهها در کنترل، مدیریت، پیشبینی و درمان CHD مؤثر خواهد بود و موجب ارتقای سلامت مبتلایان خواهد شد.
سپاسگزاری
نویسندگان مقاله از حمایتهای دانشگاه یزد در انجام این پژوهش که در راستای پایاننامه کارشناسی ارشد خانم تبریزی بوده است، تشکر و قدرانی میکنند.
حامی مالی: ندارد.
تعارض در منافع: وجود ندارد.
مشارکت نویسندگان
در ایده، نگارش و ویرایش مقاله کلیه نویسندگان مشارکت داشتند.
References:
1- Bernier P-L, Stefanescu A, Samoukovic G, Tchervenkov CI. The Challenge of Congenital Heart Disease Worldwide: Epidemiologic and Demographic Facts. Seminars in Thoracic and Cardiovascular Surgery: Pediatric Cardiac Surgery Annual 2010; 13(1): 26-34.
2- Zhao L, Chen L, Yang T, Wang T, Zhang S, Chen L, et al. Birth Prevalence of Congenital Heart Disease in China, 1980–2019: A Systematic Review and Meta-Analysis of 617 Studies. Eur J Epidemiol 2020; 35(7): 631-42.
3- Shieh JTC, Bittles AH, Hudgins L. Consanguinity and the Risk of Congenital Heart Disease. American J Med Genetics Part A 2012; 158A (5): 1236-41.
4- Zaidi S, Brueckner M. Genetics and Genomics of Congenital Heart Disease. Circ Res 2017; 120(6): 923-40.
5- Wang X, Li P, Chen S, Xi L, Guo Y, Guo A, et al. Influence of Genes and the Environment in Familial Congenital Heart Defects. Mol Med Rep 2014; 9(2): 695-700.
6- Sun R, Liu M, Lu L, Zheng Y, Zhang P. Congenital Heart Disease: Causes, Diagnosis, Symptoms, and Treatments. Cell Biochemistry and Biophysics 2015; 72(3): 857-60.
7- Zhu H, Kartiko S, Finnell R. Importance of Gene–Environment Interactions in the Etiology of Selected Birth Defects. Clinical Genetics 2009; 75(5): 409-23.
8- Bragança J, Pinto R, Silva B, Marques N, Leitão HS, Fernandes MT. Charting the Path: Navigating Embryonic Development to Potentially Safeguard against Congenital Heart Defects. J Personalized Med 2023; 13(8): 1263.
9- Blue GM, Kirk EP, Sholler GF, Harvey RP, Winlaw DS. Congenital Heart Disease: Current Knowledge About Causes and Inheritance. Med J Australia 2012; 197 (3): 155-59.
10- Lage K, Greenway SC, Rosenfeld JA, Wakimoto H, Gorham JM, Segrè AV, et al. Genetic and Environmental Risk Factors in Congenital Heart Disease Functionally Converge in Protein Networks Driving Heart Development. Proceedings of the National Academy of Sciences 2012; 109(35): 14035-40.
11- Roberts AE, Lacro RV. Genetics of Congenital Heart Disease. In Nadas' Pediatric Cardiology. 3ed. Elsevier, 2025; 55-63.
12- LaHaye S, Corsmeier D, Basu M, Bowman JL, Fitzgerald-Butt S, Zender G, et al. Utilization of Whole Exome Sequencing to Identify Causative Mutations in Familial Congenital Heart Disease. Circulation: Cardiovascular Genetics 2016; 9(4): 320-9.
13- Zahavich L, Bowdin S, Mital S. Use of Clinical Exome Sequencing in Isolated Congenital Heart Disease. Circulation: Cardiovascular Genetics 2017; 10(3): e001581.
14- Page DJ, Miossec MJ, Williams SG, Monaghan RM, Fotiou E, Cordell HJ, et al. Whole Exome Sequencing Reveals the Major Genetic Contributors to Nonsyndromic Tetralogy of Fallot. Circulation Research 2019; 124(4): 553-63.
15- Jin SC, Homsy J, Zaidi S, Lu Q, Morton S, DePalma SR, et al. Contribution of Rare Inherited and De Novo Variants In 2,871 Congenital Heart Disease Probands. Nature Genetics 2017; 49(11): 1593-601.
16- Peterlin A, Bertok S, Writzl K, Lovrečić L, Maver A, Peterlin B, et al. The Genetic Architecture of Congenital Heart Disease in Neonatal Intensive Care Unit Patients, The Experience of University Medical Centre, Ljubljana. Life 2024; 14(9): 1118.
17- Majumdar U, Yasuhara J, Garg V. In Vivo and in Vitro Genetic Models of Congenital Heart Disease. Cold Spring Harbor Perspectives in Biology 2021; 13(4): a036764.
18- Lin H, McBride KL, Garg V, Zhao MT. Decoding Genetics of Congenital Heart Disease Using Patient-Derived Induced Pluripotent Stem Cells (iPSCs). Front Cell Dev Biol 2021; 9: 630069.
19- Guo H, Liu L, Nishiga M, Cong L, Wu JC. Deciphering Pathogenicity of Variants of Uncertain Significance with CRISPR-Edited Ipscs. Trends in Genetics 2021; 37(12): 1109-23.
20- Khairy P, Ionescu-Ittu R, Mackie AS, Abrahamowicz M, Pilote L, Marelli AJ. Changing Mortality in Congenital Heart Disease. J American College of Cardiology 2010; 56(14): 1149-57.
21- Zimmerman MS, Smith AGC, Sable CA, Echko MM, Wilner LB, Olsen HE, et al. Global, Regional, and National Burden of Congenital Heart Disease: A Systematic Analysis for the Global Burden of Disease Study 2017. Lancet Child Adolescent Health 2020; 4(3): 185-200.
22- Shabana NA, Shahid SU, Irfan U. Genetic Contribution to Congenital Heart Disease (CHD). Pediatr Cardiol 2020; 41(1): 12-23.
23- Suluba E, Shuwei L, Xia Q, Mwanga A. Congenital Heart Diseases: Genetics, Non-Inherited Risk Factors, and Signaling Pathways. Egyptian J Med Human Genetics 2020; 21(1): 11.
24- Evans PR. Cardiac Anomalies in Mongolism. British Heart Journal 1950; 12(3): 258-62.
25- Nawaz K, Alifah N, Hussain T, Hameed H, Ali H, Hamayun S, et al. From Genes to Therapy: A Comprehensive Exploration of Congenital Heart Disease Through the Lens of Genetics and Emerging Technologies. Current Problems in Cardiology 2024; 49(9): 102726.
26- Moyer AJ, Gardiner K, Reeves RH. All Creatures Great and Small: New Approaches for Understanding Down Syndrome Genetics. Trends in Genetics 2021; 37(5): 444-459.
27- Irving CA, Chaudhari MP. Cardiovascular Abnormalities in Down Syndrome: Spectrum, Management and Survival Over 22 Years. Arch Dis Child 2012; 97(4): 326-30.
28- Pfitzer C, Helm PC, Rosenthal L-M, Berger F, Bauer UMM, Schmitt KRL. Dynamics in Prevalence of Down Syndrome in Children with Congenital Heart Disease. Europ J Pediatrics 2018; 177(1): 107-15.
29- Narayan P, Richter F, Morton S. Genetics and Etiology of Congenital Heart Disease. Curr Top Dev Biol 2024; 156: 297-331.
30- Obler D, Juraszek AL, Smoot LB, Natowicz MR. Double Outlet Right Ventricle: Aetiologies and Associations. J Med Genet 2008; 45(8): 481-97.
31- Qiao F, Wang Y, Zhang C, Zhou R, Wu Y, Wang C, et al. Comprehensive Evaluation of Genetic Variants Using Chromosomal Microarray Analysis and Exome Sequencing in Fetuses with Congenital Heart Defect. Ultrasound Obstet Gynecol 2021; 58 (3): 377-87.
32- Costain G, Silversides CK, Bassett AS. The Importance of Copy Number Variation in Congenital Heart Disease. NPJ Genom Med 2016; 1(1): 16031.
33- Zhao Y, Diacou A, Johnston HR, Musfee FI, McDonald-McGinn DM, McGinn D, et al. Complete Sequence of the 22q11.2 Allele in 1,053 Subjects with 22q11.2 Deletion Syndrome Reveals Modifiers of Conotruncal Heart Defects. Am J Hum Genet 2020; 106(1): 26-40.
34- Škorić-Milosavljević D, Lahrouchi N, Bosada FM, Dombrowsky G, Williams SG, Lesurf R, et al. Rare Variants in KDR, Encoding VEGF Receptor 2, Are Associated with Tetralogy of Fallot. Genetics in Medicine 2021; 23(10): 1952-1960.
35- Duran I, Tenney J, Warren CM, Sarukhanov A, Csukasi F, Skalansky M, et al. NRP1 Haploinsufficiency Predisposes to the Development of Tetralogy of Fallot. American Journal of Medical Genetics Part A 2018; 176(3): 649-56.
36- Glessner JT, Bick AG, Ito K, Homsy JG, Rodriguez-Murillo L, Fromer M, et al. Increased Frequency of De Novo Copy Number Variants in Congenital Heart Disease by Integrative Analysis of Single Nucleotide Polymorphism Array and Exome Sequence Data. Circulation Research 2014; 115(10): 884-96.
37- Soemedi R, Wilson Ian J, Bentham J, Darlay R, Töpf A, Zelenika D, et al. Contribution of Global Rare Copy-Number Variants to the Risk of Sporadic Congenital Heart Disease. Am J Human Genet 2012; 91(3): 489-501.
38- Teekakirikul P, Zhu W, Gabriel GC, Young CB, Williams K, Martin LJ, et al. Common Deletion Variants Causing Protocadherin; Deficiency Contribute to the Complex Genetics of BAV and Left-Sided Congenital Heart Disease. Human Genetics and Genomics Advances 2021; 2(3): 100037.
39- Griffin EL, Nees SN, Morton SU, Wynn J, Patel N, Jobanputra V, et al. Evidence-Based Assessment of Congenital Heart Disease Genes to Enable Returning Results in a Genomic Study. Circ Genom Precis Med 2023; 16 (2): e003791.
40- Landis BJ, Helvaty LR, Geddes GC, Lin JHI, Yatsenko SA, Lo CW, et al. A Multicenter Analysis of Abnormal Chromosomal Microarray Findings in Congenital Heart Disease. J Am Heart Assoc 2023; 12 (18): e029340.
41- Boskovski MT, Homsy J, Nathan M, Sleeper LA, Morton S, Manheimer KB, et al. De Novo Damaging Variants, Clinical Phenotypes, and Post-Operative Outcomes in Congenital Heart Disease. Circ Genom Precis Med 2020; 13(4): e002836.
42- Falsaperla R, Giacchi V, Aguglia MG, Mailo J, Longo MG, Natacci F, et al. Monogenic Syndromes with Congenital Heart Diseases in Newborns (Diagnostic Clues for Neonatologists): A Critical Analysis with Systematic Literature Review. J Pediatr Genet 2021; 10(3): 173-93.
43- Khatami M, Ghazinader D, Ahmadi F, Heidari MM, Hadadzadeh M, Namnabat M. Novel Missense Mutation in NKX2.6 Gene (C.389 G > C, Arg130Pro) As A Potentially Pathogenic Variant in Pediatric Patients with Congenital Heart Disease. Gene Reports 2023; 33: 101819.
44- Hsieh A, Morton SU, Willcox JAL, Gorham JM, Tai AC, Qi H, et al. EM-Mosaic Detects Mosaic Point Mutations that Contribute to Congenital Heart Disease. Genome Med 2020; 12(1): 42.
45- Boyle L, Wamelink MMC, Salomons GS, Roos B, Pop A, Dauber A, et al. Mutations in TKT Are the Cause of a Syndrome Including Short Stature, Developmental Delay, And Congenital Heart Defects. Am J Hum Genet. 2016; 98(6): 1235-42.
46- Pinna V, Daniele P, Calcagni G, Mariniello L, Criscione R, Giardina C, et al. Prevalence, Type, And Molecular Spectrum of NF1 Mutations in Patients with Neurofibromatosis Type 1 and Congenital Heart Disease. Genes 2019; 10(9): 675.
47- Teekakirikul P, Zhu W, Xu X, Young CB, Tan T, Smith AM, et al. Genetic Resiliency Associated with Dominant Lethal TPM1 Mutation Causing Atrial Septal Defect with High Heritability. Cell Rep Med 2022; 3(2): 100501.
48- Helle E, Córdova-Palomera A, Ojala T, Saha P, Potiny P, Gustafsson S, et al. Loss of Function, Missense, And Intronic Variants in NOTCH1 Confer Different Risks for Left Ventricular Outflow Tract Obstructive Heart Defects in Two European Cohorts. Genetic Epidemiol 2019; 43(2): 215-26.
49- Stephen J, Maddirevula S, Nampoothiri S, Burke JD, Herzog M, Shukla A, et al. Bi-allelic TMEM94 Truncating Variants Are Associated with Neurodevelopmental Delay, Congenital Heart Defects, and Distinct Facial Dysmorphism. The American Journal of Human Genetics 2018; 103(6): 948-67.
50- Heidari MM, Khatami M, Kamalipour A, Kalantari M, Movahed M, Emmamy MH, et al. Mitochondrial Mutations in Protein Coding Genes of Respiratory Chain Including Complexes IV, V, and Mt-Trna Genes Are Associated Risk Factors for Congenital Heart Disease. Excli j 2022; 21: 1306-30.
51- Khatami M, Heidari MM, Karimian N, Hadadzadeh M. Mitochondrial Mutations in tRNAGlu and Cytochrome b Genes Associated with Iranian Congenial Heart Disease. Int Cardiovasc Res J 2016; 10(4): e9815.
52- Canac R, Cimarosti B, Girardeau A, Forest V, Olchesqui P, Poschmann J, et al. Deciphering Transcriptional Networks during Human Cardiac Development. Cells 2022; 11(23): 3915.
53- Dianatpour S, Khatami M, Heidari MM, Hadadzadeh M. Novel Point Mutations of CITED2 Gene are Associated with Non-Familial Congenital Heart Disease (CHD) in Sporadic Pediatric Patients. Appl Biochem Biotechnol 2020; 190(3): 896-906.
54- Khatami M, Heidari MM, Kazeminasab F, Zare Bidaki R. Identification of A Novel Non-Sense Mutation in TBX5 Gene in Pediatric Patients with Congenital Heart Defects. J Cardiovasc Thorac Res 2018; 10(1): 41-5.
55- Tabrizi F, Khatami M, Heidari MM, Bragança J, Tatari H, Namnabat M, et al. Novel and Deleterious Nucleotide Variations in the HAND1 Gene Probably Affect Mirna Target Sites and Protein Function in Pediatric Patients with Congenital Heart Disease. Mol Biol Reports 2024; 51(1): 468.
56- Gonzalez-Teran B, Pittman M, Felix F, Thomas R, Richmond-Buccola D, Hüttenhain R, et al. Transcription Factor Protein Interactomes Reveal Genetic Determinants in Heart Disease. Cell 2022; 185(5): 794-814.e730.
57- Wu Y, Jin X, Zhang Y, Zheng J, Yang R. Genetic and Epigenetic Mechanisms in the Development of Congenital Heart Diseases. World J Pediatr Surg 2021; 4(2): e000196.
58- MacGrogan D, Münch J, de la Pompa JL. Notch and Interacting Signalling Pathways in Cardiac Development, Disease, And Regeneration. Nat Rev Cardiol 2018; 15(11): 685-704.
59- Reuter MS, Jobling R, Chaturvedi RR, Manshaei R, Costain G, Heung T, et al. Haploinsufficiency of Vascular Endothelial Growth Factor Related Signaling Genes Is Associated with Tetralogy of Fallot. Genet Med 2019; 21(4): 1001-7.
60- Williams K, Carson J, Lo C. Genetics of Congenital Heart Disease. Biomolecules 2019; 9(12): 879.
61- Hanna A, Frangogiannis NG. The Role of the TGF-β Superfamily in Myocardial Infarction. Front Cardiovasc Med 2019; 6.
62- Stefanovic S, Zaffran S. Mechanisms of Retinoic Acid Signaling during Cardiogenesis. Mech Dev 2017; 143: 9-19.
63- Nakajima Y. Retinoic Acid Signaling in Heart Development. Genesis 2019; 57 (7-8): e23300.
64- Zaffran S, Robrini NE, Bertrand N. Retinoids and Cardiac Development. J Develop Biology 2014; 2(1): 50-71.
65- Wang G, Wang B, Yang P. Epigenetics in Congenital Heart Disease. J Am Heart Assoc 2022; 11(7): e025163.
66- Coppola A, Romito A, Borel C, Gehrig C, Gagnebin M, Falconnet E, et al. Cardiomyogenesis Is Controlled by the Mir-99a/Let-7c Cluster and Epigenetic Modifications. Stem Cell Res 2014; 12(2): 323-37.
67- Linglart L, Bonnet D. Epigenetics and Congenital Heart Diseases. J Cardiovasc Dev Dis 2022; 9(6): 185.
68- Lim TB, Foo SYR, Chen CK. The Role of Epigenetics in Congenital Heart Disease. Genes (Basel) 2021; 12(3): 390.
69- Wang G, Wang B, Yang P. Epigenetics in Congenital Heart Disease. J Am Heart Assoc 2022; 11(7): e025163.
70- Wu Y, Jin X, Zhang Y, Zheng J, Yang R. Genetic and Epigenetic Mechanisms in the Development of Congenital Heart Diseases. World J Pediatr Surg 2021; 4(2): e000196.
71- Gilsbach R, Preissl S, Grüning BA, Schnick T, Burger L, Benes V, et al. Dynamic DNA Methylation Orchestrates Cardiomyocyte Development, Maturation and Disease. Nat Commun 2014; 5: 5288.
72- Asim A, Agarwal S, Panigrahi I, Saiyed N, Bakshi S. MTHFR Promoter Hypermethylation May Lead to Congenital Heart Defects in Down Syndrome. Intractable & Rare Diseases Research 2017; 6(4): 295-8.
73- Ho L, Crabtree GR. Chromatin Remodeling during Development. Nature 2010; 463(7280): 474-84.
74- Hang CT, Yang J, Han P, Cheng H-L, Shang C, Ashley E, et al. Chromatin Regulation by Brg1 Underlies Heart Muscle Development and Disease. Nature 2010; 466 (7302): 62-7.
75- Chang S, McKinsey TA, Zhang CL, Richardson JA, Hill JA, Olson EN. Histone Deacetylases 5 and 9 Govern Responsiveness of the Heart to a Subset of Stress Signals and Play Redundant Roles in Heart Development. Mol Cell Biol 2004; 24(19): 8467-76.
76- Park CY, Pierce SA, von Drehle M, Ivey KN, Morgan JA, Blau HM, et al. Sknac, A Smyd1-Interacting Transcription Factor, Is Involved in Cardiac Development and Skeletal Muscle Growth and Regeneration. Proc Natl Acad Sci USA 2010; 107(48): 20750-5.
77- Consortium EP. An Integrated Encyclopedia of DNA Elements in the Human Genome. Nature 2012; 489(7414): 57-74.
78- Chung I-M, Rajakumar G. Genetics of Congenital Heart Defects: The NKX2-5 Gene, a Key Player. Genes 2016; 7(2): 6.
79- Fotiou E, Williams S, Martin-Geary A, Robertson DL, Tenin G, Hentges KE, et al. Integration of Large-Scale Genomic Data Sources with Evolutionary History Reveals Novel Genetic Loci for Congenital Heart Disease. Circ Genom Precis Med 2019; 12(10): 442-51.
80- Nees SN, Chung WK. Genetic Basis of Human Congenital Heart Disease. Cold Spring Harbor Perspectives in Biology 2020; 12(9): a036749.
81- Touma M. Genome Regulation by Long Noncoding Rnas in Neonatal Heart Maturation and Congenital Heart Defects. J Clin and Mol Med 2020; 3(1): 1-6.
82- Dickel DE, Barozzi I, Zhu Y, Fukuda-Yuzawa Y, Osterwalder M, Mannion BJ, et al. Genome-Wide Compendium and Functional Assessment of in Vivo Heart Enhancers. Nat Commun 2016; 7: 12923.
83- Smemo S, Campos LC, Moskowitz IP, Krieger JE, Pereira AC, Nobrega MA. Regulatory Variation in a TBX5 Enhancer Leads to Isolated Congenital Heart Disease. Human Mol Genetics 2012; 21(14): 3255-63.
84- Richter F, Morton SU, Kim SW, Kitaygorodsky A, Wasson LK, Chen KM, et al. Genomic Analyses Implicate Noncoding De Novo Variants in Congenital Heart Disease. Nature Genetics 2020; 52(8): 769-77.
85- Lahm H, Jia M, Dreßen M, Wirth F, Puluca N, Gilsbach R, et al. Congenital Heart Disease Risk Loci Identified by Genome-Wide Association Study in European Patients. J Clin Invest 2021; 131(2): e141837.
86- Cordell HJ, Bentham J, Topf A, Zelenika D, Heath S, Mamasoula C, et al. Genome-Wide Association Study of Multiple Congenital Heart Disease Phenotypes Identifies a Susceptibility Locus for Atrial Septal Defect at Chromosome 4p16. Nat Genet 2013; 45(7): 822-4.
87- Agopian A, Goldmuntz E, Hakonarson H, Sewda A, Taylor D, Mitchell LE. Genome-Wide Association Studies and Meta-Analyses for Congenital Heart Defects. Circ Cardiovasc Genet 2017; 10(3): e001449
88- Yu M, Aguirre M, Jia M, Gjoni K, Cordova-Palomera A, Munger C, et al. Oligogenic Architecture of Rare Noncoding Variants Distinguishes 4 Congenital Heart Disease Phenotypes. Circ Genom Precis Med 2023;16(3): 258-66.
89- Khera AV, Chaffin M, Aragam KG, Haas ME, Roselli C, Choi SH, et al. Genome-Wide Polygenic Scores for Common Diseases Identify Individuals with Risk Equivalent to Monogenic Mutations. Nature Genetics 2018; 50(9): 1219-24.
90- Ameres SL, Zamore PD. Diversifying Microrna Sequence and Function. Nat Rev Mol Cell Biol 2013; 14(8): 475-88.
91- Fischer SEJ. RNA Interference and MicroRNA-Mediated Silencing. Curr Protoc Mol Biol 2015; 112: 26.21.21-26.21.25.
92- Diener C, Keller A, Meese E. The Mirna–Target Interactions: An Underestimated Intricacy. Nucleic Acids Res 2023; 52(4): 1544-57.
93- Sempere LF, Freemantle S, Pitha-Rowe I, Moss E, Dmitrovsky E, Ambros V. Expression Profiling of Mammalian Micrornas Uncovers a Subset of Brain-Expressed Micrornas with Possible Roles in Murine and Human Neuronal Differentiation. Genome Biol 2004; 5(3): R13.
94- Lagos-Quintana M, Rauhut R, Yalcin A, Meyer J, Lendeckel W, Tuschl T. Identification of Tissue-Specific Micrornas from Mouse. Curr Biol 2002; 12(9): 735-9.
95- Callis TE, Pandya K, Seok HY, Tang RH, Tatsuguchi M, Huang ZP, et al. Microrna-208a Is a Regulator of Cardiac Hypertrophy and Conduction in Mice. J Clin Invest 2009; 119(9): 2772-86.
96- Liu N, Williams AH, Kim Y, McAnally J, Bezprozvannaya S, Sutherland LB, et al. An Intragenic MEF2-Dependent Enhancer Directs Muscle-Specific Expression of Micrornas 1 and 133. Proc Natl Acad Sci USA 2007; 104(52): 20844-9.
97- Zhao Y, Samal E, Srivastava D. Serum Response Factor Regulates a Muscle-Specific Microrna that Targets Hand2 during Cardiogenesis. Nature 2005; 436(7048): 214-20.
98- McCarthy JJ. Microrna-206: The Skeletal Muscle-Specific Myomir. Biochim Biophys Acta 2008; 1779(11): 682-91.
99- Dueñas A, Expósito A, Aranega A, Franco D. The Role of Non-Coding Rna in Congenital Heart Diseases. J Cardiovasc Dev Dis 2019; 6(2): 15.
100- Khatami M, Ghorbani S, Adriani MR, Bahaloo S, Naeini MA, Heidari MM, et al. Novel Point Mutations in 3′-Untranslated Region of GATA4 Gene Are Associated with Sporadic Non-Syndromic Atrial and Ventricular Septal Defects. Curr Med Sci 2022; 42(1): 129-43.
101- Clowes C, Boylan M, Ridge L, Barnes E, Wright J, Hentges K. The Functional Diversity of Essential Genes Required for Mammalian Cardiac Development. Genesis 2014; 52(8): 713-37.
102- Kalayinia S, Arjmand F, Maleki M, Malakootian M, Singh CP. Micrornas: Roles in Cardiovascular Development and Disease. Cardiovasc Pathol 2021; 50: 107296.
103- Anderson KM, Anderson DM. Lncrnas at the Heart of Development and Disease. Mamm Genome 2022; 33(2): 354-65.
104- Alexanian M, Ounzain S. Long Noncoding RNAs in Cardiac Development. Cold Spring Harb Perspect Biol 2020; 12(11): a037374.
105- Martens L, Rühle F, Witten A, Meder B, Katus HA, Arbustini E, et al. A Genetic Variant Alters the Secondary Structure of the Lncrna H19 and is Associated with Dilated Cardiomyopathy. RNA Biol 2021; 18(sup1): 409-15.
106- Sweta S, Dudnakova T, Sudheer S, Baker AH, Bhushan R. Importance of Long Non-Coding Rnas in the Development and Disease of Skeletal Muscle and Cardiovascular Lineages. Front Cell Devel Biol 2019; 7: 228.
ارسال پیام به نویسنده مسئول
| بازنشر اطلاعات | |
 |
این مقاله تحت شرایط Creative Commons Attribution-NonCommercial 4.0 International License قابل بازنشر است. |
