

دوره 31، شماره 12 - ( اسفند 1402 )
جلد 31 شماره 12 صفحات 7294-7273 |
برگشت به فهرست نسخه ها


![]()
![]()
![]()
Download citation:
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:



BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
Heidari M M, Afkhami aqhda E, Tahmasebi M, Khatami M, Shaker Ardakani Z. Genetic Causes of Familial Adenomatous Polyposis (FAP), Risk Factors and Clinical Outcomes. JSSU 2024; 31 (12) :7273-7294
URL: http://jssu.ssu.ac.ir/article-1-6091-fa.html
URL: http://jssu.ssu.ac.ir/article-1-6091-fa.html
حیدری محمد مهدی، افخمی عقدا الهام، طهماسبی مریم، خاتمی مهری، شاکر اردکانی زهرا. علل ژنتیکی بیماری پولیپوز آدنوماتوز خانوادگی (FAP)، ریسکفاکتورها و پیامدهای بالینی. مجله علمي پژوهشي دانشگاه علوم پزشكي شهید صدوقی يزد. 1402; 31 (12) :7273-7294



متن کامل [PDF 1207 kb]
(655 دریافت)
| چکیده (HTML) (2144 مشاهده)
References:
1- Ditonno I, Novielli D, Celiberto F, Rizzi S, Rendina M, Ierardi E, et al. Molecular Pathways of Carcinogenesis in Familial Adenomatous Polyposis. Int J Mol Sci 2023; 24(6): 5687.
2- Half E, Bercovich D, Rozen P. Familial Adenomatous Polyposis. Orphanet J Rare Dis 2009; 4: 22.
3- Claes K, Dahan K, Tejpar S, De Paepe A, Bonduelle M, Abramowicz M, et al. The Genetics of Familial Adenomatous Polyposis (Fap) and Mutyh-Associated Polyposis (Map). Acta Gastroenterol Belg 2011; 74(3): 421-6.
4- Heidari MM, Ebrahimi F, Shaker Ardakani Z, Mirzaei M, Mirhosseini S, Khatami M. Molecular Study of Nucleotide Changes of Atpase6 and Mt-Cyb Genes in the Mitochondrial Genome of Patients with Familial Adenomatous Polyposis (Fap). The Journal of Shahid Sadoughi University of Medical Sciences 2023; 31 (5): 6632-45.
5- Afkhami E, Heidari MM, Khatami M, Ghadamyari F, Dianatpour S. Detection of Novel Mitochondrial Mutations in Cytochrome C Oxidase Subunit 1 (Cox1) in Patients with Familial Adenomatous Polyposis (Fap). Clin Transl Oncol 2020; 22(6): 908-18.
6- Heidari MM, Afkhami E, Khatami M, Farzaneh G. Genetic Analysis of D-Loop Region of Mitochondrial Dna Sequence in Iranian Patients with Familial Adenomatous Polyposis (Fap): A Case-Control Study. Journal of Rafsanjan University of Medical Sciences 2023; 21(12): 1307-22.
7- Shaker Ardakani Z, Heidari MM, Khatami M, Bitaraf Sani M. Association of Pathogenic Missense and Nonsense Mutations in Mitochondrial Coii Gene with Familial Adenomatous Polyposis (Fap). Int J Mol Cell Med 2020; 9(4): 255-65.
8- Parker TW, Neufeld KL. Apc Controls Wnt-Induced Β-Catenin Destruction Complex Recruitment in Human Colonocytes. Sci Rep 2020; 10(1): 2957.
9- Hankey W, Frankel WL, Groden J. Functions of the Apc Tumor Suppressor Protein Dependent and Independent of Canonical Wnt Signaling: Implications for Therapeutic Targeting. Cancer Metastasis Rev 2018; 37(1): 159-72.
10- Pino MS, Chung DC. The Chromosomal Instability Pathway in Colon Cancer. Gastroenterology 2010; 138(6): 2059-72.
11- Kapitanović S, Cacev T, Radosević S, Spaventi S, Spaventi R, Pavelić K. Apc Gene Loss of Heterozygosity, Mutations, E1317q, and I1307k Germ-Line Variants in Sporadic Colon Cancer in Croatia. Exp Mol Pathol 2004; 77(3): 193-200.
12- Nielsen M, Hes FJ, Nagengast FM, Weiss MM, Mathus-Vliegen EM, Morreau H, et al. Germline Mutations in Apc and Mutyh are Responsible for the Majority of Families with Attenuated Familial Adenomatous Polyposis. Clin Genet 2007; 71(5): 427-33.
13- Balmaña J, Balaguer F, Cervantes A, Arnold D; ESMO Guidelines Working Group. Familial Risk-Colorectal Cancer: Esmo Clinical Practice Guidelines. Ann Oncol 2013; 24 Suppl 6: vi73-80.
14- Ghadamyari F, Heidari MM, Zeinali S, Khatami M, Merat S, Bagherian H, Rejali L. Mutational Screening through Comprehensive Bioinformatics Analysis to Detect Novel Germline Mutations in the Apc Gene in Patients with Familial Adenomatous Polyposis (Fap). J Clin Lab Anal 2021; 35(5): e23768.
15- Rowan AJ, Lamlum H, Ilyas M, Wheeler J, Straub J, Papadopoulou A, et al. APC Mutations in Sporadic Colorectal Tumors: A Mutational "Hotspot" and Interdependence of the "Two Hits" . Proc Natl Acad Sci U S A 2000; 97(7): 3352-7.
16- De Queiroz Rossanese LB, De Lima Marson FA, Ribeiro JD, Coy CS, Bertuzzo CS. Apc Germline Mutations in Families with Familial Adenomatous Polyposis. Oncol Rep 2013; 30(5): 2081-8.
17- Heinen CD. Genotype to Phenotype: Analyzing the Effects of Inherited Mutations in Colorectal Cancer Families. Mutat Res 2010; 693(1-2): 32-45.
18- Schneikert J, Behrens J. The Canonical Wnt Signalling Pathway and its Apc Partner in Colon Cancer Development. Gut 2007; 56(3): 417-25.
19- Dallosso AR, Jones S, Azzopardi D, Moskvina V, Al-Tassan N, Williams GT, et al. The Apc Variant P.Glu1317gln Predisposes to Colorectal Adenomas by a Novel Mechanism of Relaxing the Target for Tumorigenic Somatic Apc Mutations. Hum Mutat 2009; 30(10): 1412-8.
20- Disciglio V, Forte G, Fasano C, Sanese P, Lepore Signorile M, De Marco K, et al. Apc Splicing Mutations Leading to in-Frame Exon 12 or Exon 13 Skipping are Rare Events in Fap Pathogenesis and Define the Clinical Outcome. Genes(Basel) 2021; 12(3): 353.
21- Schwab AL, Tuohy TM, Condie M, Neklason DW, Burt RW. Gonadal Mosaicism and Familial Adenomatous Polyposis. Fam Cancer 2008; 7(2): 173-77.
22- Aitchison A, Hakkaart C, Day RC, Morrin HR, Frizelle FA, Keenan JI. Apc Mutations are Not Confined to Hotspot Regions in Early-Onset Colorectal Cancer. Cancers(Basel) 2020; 12(12): 3829.
23- Fearnhead NS, Britton MP, Bodmer WF. The Abc of Apc. Hum Mol Genet 2001; 10(7): 721-33.
24- Talseth-Palmer BA. The Genetic Basis of Colonic Adenomatous Polyposis Syndromes. Hered Cancer Clin Pract 2017; 15: 5.
25- Sant V, Reich E, Khanna L, Cao W, Kornacki S, Grucela A.. Attenuated Familial Adenomatous Polyposis (Afap) in a Patient Associated with a Novel Mutation in Apc. BMJ Case Rep 2019; 12(11): e231232.
26- Ibrahim A, Barnes DR, Dunlop J, Barrowdale D, Antoniou AC, Berg JN. Attenuated Familial Adenomatous Polyposis Manifests as Autosomal Dominant Late-Onset Colorectal Cancer. Eur J Hum Genet 2014; 22(11): 1330-3.
27- Latchford A, Volikos E, Johnson V, Rogers P, Suraweera N, Tomlinson I, et al. Apc Mutations in Fap-Associated Desmoid Tumours are Non-Random but Not 'Just Right'. Hum Mol Genet 2007; 16(1): 78-82.
28- Mankaney G, Leone P, Cruise M, LaGuardia L, O'Malley M, Bhatt A, et al. Gastric Cancer in Fap: A Concerning Rise in Incidence. Fam Cancer 2017; 16(3): 371-6.
29- Campos FG, Martinez CAR, Bustamante Lopez LA, Kanno DT, Nahas SC, Cecconello I. Advanced Duodenal Neoplasia and Carcinoma in Familial Adenomatous Polyposis: Outcomes of Surgical Management. J Gastrointest Oncol 2017; 8(5): 877-84.
30- Groves CJ, Saunders BP, Spigelman AD, Phillips RK. Duodenal Cancer in Patients with Familial Adenomatous Polyposis (Fap): Results of a 10 Year Prospective Study. Gut 2002; 50(5): 636-41.
31- Bülow S, Björk J, Christensen IJ, Fausa O, Järvinen H, Moesgaard F, et al. Duodenal Adenomatosis in Familial Adenomatous Polyposis. Gut 2004; 53(3): 381-6.
32- Deibert B, Ferris L, Sanchez N, Weishaar P. The Link Between Colon Cancer and Congenital Hypertrophy of the Retinal Pigment Epithelium (Chrpe). Am J Ophthalmol Case Rep 2019; 15: 100524.
33- Wijn MA, Keller JJ, Giardiello FM, Brand HS. Oral and Maxillofacial Manifestations of Familial Adenomatous Polyposis. Oral Dis 2007; 13(4): 360-5.
34- Groen EJ, Roos A, Muntinghe FL, Enting RH, de Vries J, Kleibeuker JH, et al. Extra-Intestinal Manifestations of Familial Adenomatous Polyposis. Ann Surg Oncol 2008; 15(9): 2439-50.
35- Vasen HF, Möslein G, Alonso A, Aretz S, Bernstein I, Bertario L, et al. Guidelines for the Clinical Management of Familial Adenomatous Polyposis (Fap). Gut 2008; 57(5): 704-13.
36- Abdullah Suhaimi SN, Nazri N, Nani Harlina ML, Md Isa N, Muhammad R. Familial Adenomatous Polyposis-Associated Papillary Thyroid Cancer. Malays J Med Sci 2015; 22(4): 69-72.
37- Farinella E, Soobrah R, Phillips RK, Clark SK. Familial Adenomatous Polyposis (Fap) and Gender. Does Gender Influence the Genetic Transmission of Fap? Fam Cancer 2010; 9(3): 405-6.
38- Waller A, Findeis S, Lee MJ. Familial Adenomatous Polyposis. J Pediatr Genet 2016; 5(2): 78-83.
39- Leppert M, Burt R, Hughes JP, Samowitz W, Nakamura Y, Woodward S, et al. Genetic Analysis of an Inherited Predisposition to Colon Cancer in a Family with a Variable Number of Adenomatous Polyps. N Engl J Med 1990; 322(13): 904-8.
40- Ripa R, Bisgaard ML, Bülow S, Nielsen FC. De Novo Mutations in Familial Adenomatous Polyposis (Fap). Eur J Hum Genet 2002; 10(10): 631-7.
41- Torrezan GT, da Silva FC, Santos EM, Krepischi AC, Achatz MI, Aguiar S Jr, et al. Mutational Spectrum of the Apc and Mutyh Genes and Genotype-Phenotype Correlations in Brazilian Fap, Afap, and Map Patients. Orphanet J Rare Dis 2013; 8: 54.
42- Newton KF, Mallinson EK, Bowen J, Lalloo F, Clancy T, Hill J, et al. Genotype-Phenotype Correlation in Colorectal Polyposis. Clin Genet 2012; 81(6): 521-31.
43- Nieuwenhuis MH, Vasen HF. Correlations between Mutation Site in Apc and Phenotype of Familial Adenomatous Polyposis (Fap): A Review of the Literature. Crit Rev Oncol Hematol 2007; 61(2): 153-61.
44- Dinarvand P, Davaro EP, Doan JV, Ising ME, Evans NR, Phillips NJ, et al. Familial Adenomatous Polyposis Syndrome: An Update and Review of Extraintestinal Manifestations. Arch Pathol Lab Med 2019; 143(11): 1382-98.
45- Renkonen ET, Nieminen P, Abdel-Rahman WM, Moisio AL, Järvelä I, Arte S, et al. Adenomatous Polyposis Families That Screen Apc Mutation-Negative By Conventional Methods Are Genetically Heterogeneous. J Clin Oncol 2005; 23(24): 5651-9.
46- Aihara H, Kumar N, Thompson CC. Diagnosis, Surveillance, and Treatment Strategies for Familial Adenomatous Polyposis: Rationale and Update. Eur J Gastroenterol Hepatol 2014; 26(3): 255-62.
47- Wang D, Liang S, Zhang X, Dey SK, Li Y, Xu C, et al. Targeted Next-Generation Sequencing Approach for Molecular Genetic Diagnosis of Hereditary Colorectal Cancer: Identification of a Novel Single Nucleotide Germline Insertion in Adenomatous Polyposis Coli Gene Causes Familial Adenomatous Polyposis. Mol Genet Genomic Med 2019; 7(1): e00505.
48- Herzig D, Hardiman K, Weiser M, You N, Paquette I, Feingold DL, et al. The American Society of Colon and Rectal Surgeons Clinical Practice Guidelines for the Management of Inherited Polyposis Syndromes. Dis Colon Rectum 2017; 60(9): 881-94.
49- Tajika M, Tanaka T, Oonishi S, Yamada K, Kamiya T, Mizuno N, et al. Endoscopic Management of Adenomas in the Ileal Pouch and the Rectal Remnant after Surgical Treatment in Familial Adenomatous Polyposis. J Clin Med 2022; 11(12): 3562.
50- Hyer W, Cohen S, Attard T, Vila-Miravet V, Pienar C, Auth M, et al. Management of Familial Adenomatous Polyposis in Children and Adolescents: Position Paper from the Espghan Polyposis Working Group. J Pediatr Gastroenterol Nutr 2019; 68(3): 428-41.
51- Tudyka VN, Clark SK. Surgical Treatment in Familial Adenomatous Polyposis. Ann Gastroenterol 2012; 25(3): 201-6.
52- Anele CC, Xiang J, Martin I, Hawkins M, Man R, Clark SK, et al. Regular Endoscopic Surveillance and Polypectomy is Effective in Managing Rectal Adenoma Progression Following Colectomy and Ileorectal Anastomosis in Patients with Familial Adenomatous Polyposis. Colorectal Dis 2022; 24(3): 277-83.
53- Thiruvengadam SS, Lopez R, O'Malley M, LaGuardia L, Church JM, Kalady M, et al. Spigelman Stage Iv Duodenal Polyposis Does Not Precede Most Duodenal Cancer Cases in Patients with Familial Adenomatous Polyposis. Gastrointest Endosc 2019; 89(2): 345-54. e2.
54- Lopez-Ceron M, van den Broek FJ, Mathus-Vliegen EM, Boparai KS, van Eeden S, Fockens P, et al. The Role of High-Resolution Endoscopy and Narrow-Band Imaging in the Evaluation of Upper Gi Neoplasia in Familial Adenomatous Polyposis. Gastrointest Endosc 2013; 77(4): 542-50.
55- Campos FG, Sulbaran M, Safatle-Ribeiro AV, Martinez CA. Duodenal Adenoma Surveillance in Patients with Familial Adenomatous Polyposis. World J Gastrointest Endosc 2015; 7(10): 950-9.
56- Belfiore A, Ciniselli CM, Signoroni S, Gariboldi M, Mancini A, Rivoltini L, et al. Preventive Anti-Inflammatory Diet to Reduce Gastrointestinal Inflammation in Familial Adenomatous Polyposis Patients: A Prospective Pilot Study. Cancer Prev Res (Phila) 2021; 14(10): 963-72.
57- Nieuwenhuis MH, Mathus-Vliegen EM, Baeten CG, Nagengast FM, van der Bijl J, van Dalsen AD, et al. Evaluation of Management of Desmoid Tumours Associated with Familial Adenomatous Polyposis in Dutch Patients. Br J Cancer 2011; 104(1): 37-42.
58- Iwama T, Akasu T, Utsunomiya J, Muto T. Does a Selective Cyclooxygenase-2 Inhibitor (Tiracoxib) Induce Clinically Sufficient Suppression of Adenomas in Patients with Familial Adenomatous Polyposis? A Randomized Double-Blind Placebo-Controlled Clinical Trial. Int J Clin Oncol 2006; 11(2): 133-9.
59- Burke CA, Dekker E, Lynch P, Samadder NJ, Balaguer F, Hüneburg R,, et al. Eflornithine Plus Sulindac for Prevention of Progression in Familial Adenomatous Polyposis. N Engl J Med 2020; 383(11): 1028-39.
60- Lynch PM, Burke CA, Phillips R, Morris JS, Slack R, Wang X, et al. An International Randomised Trial of Celecoxib Versus Celecoxib Plus Difluoromethylornithine in Patients with Familial Adenomatous Polyposis. Gut 2016; 65(2): 286-295.
61- Dovizio M, Tacconelli S, Ricciotti E, Bruno A, Maier TJ, Anzellotti P, et al. Effects of Celecoxib on Prostanoid Biosynthesis and Circulating Angiogenesis Proteins in Familial Adenomatous Polyposis. J Pharmacol Exp Ther 2012; 341(1): 242-50.
62- Aelvoet AS, Buttitta F, Ricciardiello L, Dekker E. Management of Familial Adenomatous Polyposis and Mutyh-Associated Polyposis; New Insights. Best Pract Res Clin Gastroenterol 2022; 58-59: 101793.
63- Kantor M, Sobrado J, Patel S, Eiseler S, Ochner C. Hereditary Colorectal Tumors: A Literature Review on Mutyh-Associated Polyposis. Gastroenterol Res Pract 2017; 2017: 8693182.
64- Rashid M, Fischer A, Wilson CH, Tiffen J, Rust AG, Stevens P, et al. Adenoma Development in Familial Adenomatous Polyposis and Mutyh‐Associated Polyposis: Somatic Landscape and Driver Genes. J Pathol 2016; 238(1): 98-108.
65- Disel U, Sivakumar S, Pham T, Fleischmann Z, Anu RI, Sokol ES, et al. Increased Kras G12c Prevalence, High Tumor Mutational Burden, and Specific Mutational Signatures are Associated with Mutyh Mutations: A Pan-Cancer Analysis. Oncologist 2023: oyad230.
66- Toboeva MK, Shelygin YA, Frolov SA, Kuzminov MA, Tsukanov AS. Mutyh-Associated Polyposis. Ter Arkh 2019; 91(2): 97-100.
67- Out AA, Tops CM, Nielsen M, Weiss MM, van Minderhout IJ, Fokkema IF, et al. Leiden Open Variation Database of the Mutyh Gene. Hum Mutat 2010; 31(11): 1205-15.
68- Moisio AL, Järvinen H, Peltomäki P. Genetic and Clinical Characterisation of Familial Adenomatous Polyposis: A Population Based Study. Gut 2002; 50(6): 845-50.
69- Knudsen A, Bülow S, Tomlinson I, Möslein G, Heinimann K, Christensen I, et al. Attenuated Familial Adenomatous Polyposis: Results from an International Collaborative Study. Colorectal Dis 2010; 12(10Online): e243-9.
70- Grover S, Kastrinos F, Steyerberg EW, Cook EF, Dewanwala A, Burbidge LA, et al. Prevalence and Phenotypes of Apc and Mutyh Mutations in Patients with Multiple Colorectal Adenomas. Jama 2012; 308(5): 485-92.
71- Guarinos C, Juárez M, Egoavil C, Rodríguez-Soler M, Pérez-Carbonell L, Salas R, et al. Prevalence and Characteristics of Mutyh-Associated Polyposis in Patients with Multiple Adenomatous and Serrated Polyps. Clin Cancer Res 2014; 20(5): 1158-68.
72- Seguí N, Navarro M, Pineda M, Köger N, Bellido F, González S, et al. Exome Sequencing Identifies Mutyh Mutations in a Family with Colorectal Cancer and an Atypical Phenotype. Gut 2015; 64(2): 355-6.
73- Morak M, Heidenreich B, Keller G, Hampel H, Laner A, de la Chapelle A, et al. Biallelic Mutyh Mutations Can Mimic Lynch Syndrome. Eur J Hum Genet 2014; 22(11): 1334-7.
74- Win AK, Hopper JL, Jenkins MA. Association between Monoallelic Mutyh Mutation and Colorectal Cancer Risk: A Meta-Regression Analysis. Fam Cancer 2011; 10(1): 1-9.
75- Theodoratou E, Campbell H, Tenesa A, Houlston R, Webb E, Lubbe S, et al. A Large-Scale Meta-Analysis to Refine Colorectal Cancer Risk Estimates Associated with Mutyh Variants. Br J Cancer 2010; 103(12): 1875-84.
76- Vogt S, Jones N, Christian D, Engel C, Nielsen M, Kaufmann A, et al. Expanded Extracolonic Tumor Spectrum in Mutyh-Associated Polyposis. Gastroenterology 2009; 137(6): 1976-85. e1-10
77- Nielsen M, Morreau H, Vasen HF, Hes FJ. Mutyh-Associated Polyposis (Map). Crit Rev Oncol Hematol 2011; 79 (1): 1-16.
78- Pitroski CE, Cossio SL, Koehler-Santos P, Graudenz M, Prolla JC, Ashton-Prolla P. Frequency of the Common Germline Mutyh Mutations P. G396d and P. Y179c in Patients Diagnosed with Colorectal Cancer in Southern Brazil. Int J Colorectal Dis 2011; 26(7): 841-6.
متن کامل: (3608 مشاهده)
مقدمه
پولیپوز آدنوماتوز خانوادگی (FAP) یک سندرم ارثی است که از طریق پیشرفت آدنوماهای متعدد در کولورکتوم و افزایش ریسک ابتلا به سرطان کولورکتال (CRC) و همچنین وجود علایمی در بافتهایی غیر از بافت کولون تظاهر مییابد. جهشهای ژرمینال ژن پولیپوز آدنوماتوز کولی (APC) در ابتدا در سال 1911 بهعنوان عامل بیماری FAP و با الگوی وراثتی اتوزومال غالب توصیف شد (1,2). از آن بهبعد، شواهد متعددی از جمله: یافتههای پاتوفیزیولوژی، ژنتیکی، فنوتیپیکی و علایم بالینی منجر به ابداع روشهای پیشگیرانه مناسبی در بیماران شده است. در سال 2002، ژن MutYH، بهعنوان یکی دیگر از ژنهای دخیل در پولیپوز شناسایی شد و مشخص گردید که جهشهای دوآللی آن، منجر به ایجاد FAP، با الگوی وراثتی اتوزومال مغلوب میشود که معمولا از آن بهعنوان پولیپوز وابسته به MutYH، (MAP) یاد میگردد (3). با اینحال، 20 تا 35% موارد نوظهور (Denovo) FAP، بدون شواهد بالینی یا ژنتیکی در والدین بیمار گزارش میشوند. به همین دلیل، با توجه به افزایش خطر بدخیمی، پروتکلهای غربالگری ژنتیکی برای بیماران مبتلا به FAP پیشنهاد شده است. نتایج تحقیقات قبلی ما در بیماران FAP نیز نقش پاتوژنز جهشهای ژنهای میتوکندری، علاوه بر ناهنجاریهای ژنوم هستهای، را در بیماران خانوادگی و تکگیر تایید کرد (7-4). با اینحال، در مقاله حاضر، تمرکز بر تغییرات ژنتیکی در ژنوم هستهای دخیل در فرایند بیماریزایی FAP است.
FAP وابسته به APC (APC-FAP): FAPوابسته به APC (OMIM#175100) یک بیماری ارثی با الگوی اتوزومال غالب است که از طریق گسترش چندین آدنوما در سرتاسر کولورکتوم، تظاهر مییابد. کمتر از 1% از تمام سرطانهای کولون ( (CRCرا این نوع FAP تشکیل میدهد که شیوع آن یک در هر 7000 تا 10000 نفر بوده و شایعترین پولیپوز دستگاه گوارش محسوب میشود (2).
ژن APC : APC یک ژن سرکوبگر توموری است که در موقعیت کروموزومی 5q21-22 قرار گرفته است. این ژن دارای 15 اگزون است که اگزون 15 آن، به تنهایی 75% از توالی کدکننده را شامل می¬شود و بهعنوان اصلیترین اگزون مورد هدف جهشهای ژرمینال و سوماتیک به حساب میآید. ژن APC کدکننده پروتئینی با 2843 آمینواسید (kDa310) میباشد که در مسیر سیگنالینگ Wnt بهطور مستقیم نقش دارد (8). این پروتئین چندعملکردی، حاوی چندین ایزوفرم، موتیف و دامینهای آمینواسیدی در سلول میباشد که به آن اجازه میدهد تا با مولکولهای متعدد دیگر تعامل داشته باشد (شکل 1). پروتئین APC بهعنوان یک سرکوبکننده توموری، خود از طریق تنظیم منفی انکوپروتئین-catenin β تنظیم میشود. پروتئین APC منجر به یوبیکوییتینهشدن(Ubiquitination) و تکهتکه شدن catenin –β میگردد، بنابراین در غیاب آن،catenin –β در هسته تجمع مییابد و با فاکتورهایی برهم کنش میکند که در تنظیم نسخهبرداری ژنهای درگیر در سیکل سلولی، تکثیر، تمایز، مهاجرت و آپوپتوز نقش دارند (9). علاوه بر این، APC از طریق تثبیت میکروتوبولها، منجر به افزایش پایداری کروموزومها نیز میگردد، در نتیجه غیرفعال ساختنAPC موجب تفرق ناصحیح یا عدم تفرق صحیح کروموزومها و نقص در فرآیند میتوز می¬شود (10). در اکثر موارد، FAP به دلیل جهشهای ژرمینال در ژن APC ، ایجاد میشود. افرادی با یک جهش ژرمینال در ژن APC دارای چندین آدنوما در ناحیه کولورکتوم هستند که این علایم، به دلیل غیر فعالسازی آلل سالم در اثر بروز جهشهای سوماتیک در ژن APC یا از دستدادن حالت هتروزیگوسیتی (LOH) در این لوکوس ایجاد میشوند (11). اخیرا در یک مطالعه تجربی، در 80% از افراد مبتلایی که دارای بیش از 1000 آدنوما بودند، در بیش از نیمی از افرادیکه دارای 100 تا 999 آدنوما بودند، در 10% از افراد مبتلا با 20 تا 99 آدنوما و در 5% از بیماران با 10 تا 19 آدنوما، جهش در ژن APC مشاهده گردید (12). هر چند که FAP نوعی بیماری با نحوه توارث اتوزومال غالب است، اما بیش از 25% از مبتلایان، دارای جهشهای ژرمینال از نوع نوظهور (denovo) هستند (13). از زمان شناسایی ژن APC، بیش از 1100 جهش ژرمینال بیماریزا (پاتوژن) در این ژن گزارش شده است. اکثر این جهشها از نوع تغییرات نوکلئوتیدی کوتاهکننده (Truncating) هستند که شامل: جهشهای بیمعنی (28%)، اضافهشدنهای کوچک نوکلئوتیدی (10%) یا حذفهای کوچک (46%) بوده و منجر به تولید پروتئین ناقص میشوند (14,15). همچنین جهشهای بدمعنی (3%) و تغییرات بزرگی (مانند: حذفهای تک اگزونی یا چند اگزونی و مضاعفشدگیهای نوکلئوتیدی) (13%) نیز با وجود اینکه نادر هستند، اما در بیماران گزارش شدهاند (16). در ذیل به چند مورد از چنین جهشهای مهمی در ژن APC اشاره میشود:
1) جهشهای کوتاهکننده: همانطور که ذکر شد، اکثر جهشهای ژن APC که مرتبط با بیماری FAP هستند، منجر به تولید پروتئین ناقصی میشوند که عمدتا ناشی از تغییر در چارچوب خواندنی یا یک جهش بیمعنی میباشد. جهشهای C>T، از متداولترین جهشهای بیمعنی در این ژن هستند. مشخص شده است که اکثر جهشهای ژرمینال در ژن APC در ناحیه ′5 ژن رخ میدهند که منجر به حذف اکثر تکرارهای اسیدآمینهای میشوند که در تنظیم ژنی در سطح β- کاتنین، و تکرارهای SAMP که در باندینگ آکسین درگیرند، نقش دارند (شکل A1) (17,18).
2) جهشهای بدمعنی: در تحقیقات گذشته، بیش از 60 جهش بدمعنی مختلف، که همگی واریانت ژنی محسوب شده و با وجود پایینبودن میزان شیوع، بهعنوان جهشهایی که بهصورت بالقوه بیماریزا هستند، توصیف شدهاند. Ile1307Lys و Glu1317Gln دو نوع از فراوانترین واریانتهای بدمعنی اند که تاکنون در این ژن گزارش شدهاند. هرچندکه واریانت Ile1307Lys که در 6% از یهودیان اشکنازی وجود دارد، منجر به فنوتیپ پولیپوز نمیشود، اما موجب افزایش 10 تا 20 درصدی ریسک ابتلا به CRC میگردد. واریانت Glu1317Gln نیز با یک ریسک متوسط برای خطر آدنوما و CRC همراه است (19).
3) جهشهای پیرایشگر (Splicing) و تغییرات بزرگ (Gross): این نوع جهشها موجب تغییر در الگوی پیرایش ژن و حذفشدگیها یا مضاعفشدگیهای نوکلئوتیدی میشوند. دادههای اخیر نیز حاکی از این موضوع هستند که بهویژه تغییرات ژنی بزرگ، بر روی پروموتور نواحی کدینگ موثر بوده و در بیش از 20% از خانوادههای FAP گزارش شده است (20).
4) جهشهای نوظهور و موزاییسم رده سلولهای زاینده: بسیاری از جهشهای ژرملاین در ژن APC، به ارث میرسند، اما میتوانند در فرد بیماری حتی بدون سوابق خانوادگی نیز به صورت جهشهای نوظهور، رخ بدهد که در بین 11% تا 25% از کل مبتلایان به FAP دیده میشوند. نرخ برآورد جهشهای نوظهور، بین 106× 9-4 جهش/گامت/ نسل میباشد و میزان جهشزایی آن طی اووژنز و اسپرماتوژنز برابر است. درصد قابلتوجهی از جهشهای نوظهور، به شکل موزاییک بهوجود میآیند که تنها بر روی یک رده از سلولهای فرد مبتلا تاثیر میگذارند (طبق برآوردها، یک پنجم از جهشهای نوظهور در بیماری FAP از نوع موزاییک هستند) (21).
5) نقاط داغ (Hotspots) جهش: نقاط داغ جهش در ژن APC در قسمت ′5 اگزون 15 و در کدونهای 1309 و 1061 واقع شدهاند که تقریبا 11% تا 17% از تمام جهشهای ژرمینال را تشکیل میدهند. همچنین، بهدلیل تجمع جهشها از کدون 1250 تا 1464، از این ناحیه بهعنوان ناحیه خوشهای جهش (MCR) یاد میکنند (شکل 1) (22). نوع جهش ژرمینال در ژن APC، ماهیت ضربه دوم را برای این ژن مشخص میکند. در صورتیکه جهش ژرمینال، مابین کدونهای 1194 و 1392 اتفاق بیفتد، به احتمال خیلی قوی، ضربه دوم بهصورت فقدان آللی در این ژن بروز مییابد. درمقابل، اگر جهش ژرمینال در خارج از این ناحیه رخ دهد، ضربه دوم به احتمال زیاد، یک جهش ناقصکننده در MCR است (23).
فنوتیپها: FAP کلاسیک و FAP تضعیفشده (AFAP): مطابق با تعداد پولیپها و سن فرد، دو نوع فنوتیپ اصلی برای بیماری FAP بیان میشود (جدول 1):
FAP کلاسیک: از طریق وجود صدها تا هزاران پولیپ آدنوماتوز در سرتاسر کولون و رکتوم مشخص میشود. معمولاً در زمان نوجوانی، پولیپها در رکتوسیگموئید در اندازههای کوچک، شناسایی میشوند و پس از آن اندازه و تعداد آنها افزایش مییابد. آدنوماها تقریبا در نیمی از بیماران از 15 سالگی و در 95% از بیماران در سن 35 سالگی رشد میکنند. کاملا بدیهی است که سن ابتلا به CRC از طریق جهش ژرمینال، پایینتر از سن ابتلا به CRCایی است که بهنحوه تکگیر (Sporadic) بروز مییابد (میانگین سنی مبتلایان 35 سال)، و بهندرت در سن قبل از 20 سالگی رخ میدهد (24).
AFAP: این نوع FAP، حالتی خفیفتر از FAP کلاسیک میباشد که از طریق کاهش تعداد پولیپها (100-10)، افزایش سن ابتلا به بیماری در یک فرد، توزیع پولیپها بیشتر در سمت راست (right-sided) و کاهش سن ابتلا به CRC (بیش از7%) شناسایی میشود (25).
تعریف بالینی AFAP یکی از موضوعات بحثبرانگیز میباشد و میبایست در فرد بیمار، درحدود 99-10 آدنوما مورد بررسی قرار گیرد، هرچندکه تشخیص دقیق یک بیمار، بسیار دشوار میباشد. پیشرفت پولیپ، در بیماری AFAP که وابسته به ژن APC است، مشابه ایجاد پولیپ در بیماری MAP و یا حتی بیماری FAPای است که به شکل تکگیر ایجاد میشود. بررسی چند تن از اعضای خانواده بیمار، میتواند تعیینکننده فنوتیپ FAP باشد (26).
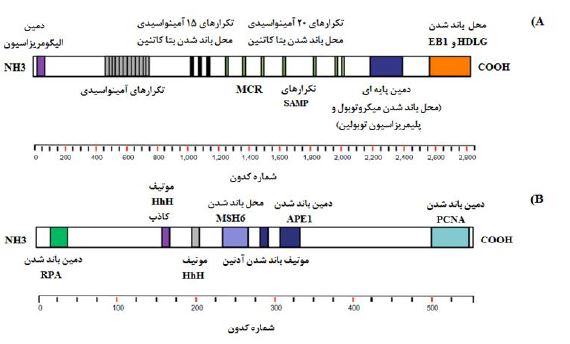
شکل 1: دامینهای عملکردی پروتئین MutYHو APC. A) پروتئین APC شامل یک دامین الیگومریزاسیون و یک منطقه armadillo در انتهای N، تعدادی از تکرارهای 15 و 20 آمینواسیدی در قسمت مرکزی آن و یک انتهای C است که شامل: یک دامین پایهای و محلهای باندشدن پروتئین EB1 و پروتئین دیسک انسانی بزرگ (HDLG) میباشد. B): پروتئین MutYH و دامینهای مختلف آن. اختصارات: پروتئین همانندسازی A (RPA)، ساختار مارپیچ-سنجاق سری-مارپیچ (HhH)، اندونوکلئاز آپورینیک1 (APE1)، آنتیژن هستهای تکثیر سلولی (PCNA)، ناحیه خوشهای جهش (MCR).
جدول 1: فنوتیپهای بالینی FAP وابسته به APC و پولیپوز وابسته به MutYH
.JPG)
FAP: پولیپوز آدنوماتوز خانوادگی، CRC: سرطان کولورکتال، AFAP: پولیپوز آدنوماتوز خانوادگی تضعیفشده، MAP: پولیپوز آدنوماتوز وابسته به MutYH، SPS: سندرم پولیپوز دندانهای، MMR : ترمیم جفت باز اشتباه
علایمی غیر از کولون: در بسیاری از بیماران مبتلا به FAP، علاوه بر کولون، بافتهای دیگر نیز درگیر میشوند و علائمی همچون پولیپهای معده و دوازدهه، تومورهای دسموئیدی (DT) و تیروئیدی و تومورهای مغزی، استخوانی (استئوما)، هایپرتروفی مادرزادی اپیتلیوم رنگدانه شبکیه، دندانهای غیرعادی و کیستهای اپیدرموئید، نیز بروز مییابند (27).
پولیپهای معده - دوازدهه: متداولترین تظاهراتی که به غیر از کولون، در این بیماران بروز پیدا میکنند، پولیپهای معده - رودهای فوقانی هستند. آنها در نواحی شکم، دوازدهه و پریآمپولار قرار میگیرند. پولیپهای معدی معمولاً از نوع پولیپهای غددی فاندیک (FGP) خوشخیم بوده و در 20% تا 84% از مبتلایان ایجاد میشوند. با این وجود، FGPهای وابسته به FAP بهطور مرسوم، بهعنوان غیرنئوپلازی در نظر گرفته میشوند و معمولاً بدون نیاز به جراحی میباشند، تاکنون مواردی از جمله دیسپلازی با درجه بالا و کارسینومای معده که ناشی از FGP بوده، در مبتلایان به FAP گزارش شده است (28,29). پولیپهای آدنوماتوی معده، در حدود 10% از پولیپهای معده حضور دارند و زمانی تشخیص داده میشوند که در محل حفرات گوارشی، به تعداد زیاد دیده شوند. علیرغم پتانسیل بدخیمی FGP، دیسپلازی معده، آدنوما و کارسینومای معده، در مبتلایان بسیار نادر میباشد (بروز کمتر از 1%). بعد از کولورکتوم، دومین و رایجترین ناحیه برای پولیپها، دوازدهه است. آدنوماهای دوازدهه در اکثر بیماران مبتلا به FAP، با ریسک تقریبی 100% ایجاد میشوند. بخشهای دوم و سوم دوازدهه، مخصوصا ناحیه پریآمپولار، دارای استعداد قابلتوجهی برای ایجاد پولیپ میباشند. این الگو احتمالا به دلیل قرارگرفتن مخاط دوازدهه در معرض اسیدهای صفراوی است که بیانگر نقش این ترکیبات در کارسینومای دوازدهه میباشد. سرطان دوازدهه با یک ریسک فزایندهی 5%، دومین علت مرگ و میر ناشی از سرطان، در بیماران مبتلا به FAP است (30,31).
نشانههای غیرکولون: هر دو نشانههای بدخیم و خوشخیم غیرکولون، در بیماران FAP شایع است. هایپرتروفی مادرزادی اپیتلیوم رنگدانه شبکیه (CHRPE) شایعترین نشانههای غیرکولون در بیماران FAP میباشد (80%-70%) که بهصورت منحنی خاکستری - قهوهای مایل به سیاه، یا ضایعات بیضیشکل در شبکیه چشم بهنظر میرسند، اما اینکه چه نوع مشکلات بالینی را ایجاد کنند، هنوز بهدرستی شناخته شده نیست. کیستهای اپیدرموئید (50%) و فیبروما (50%-25%)، بهعنوان شایعترین ضایعات زیرپوستی هستند که ممکن است موجب مشکلات ظاهری بیمار شوند. سایر نشانههای خوشخیم شامل: ناهنجاریهای دندانی (90%-79%)، پوکی استخوان (90%-50%) و تومورهای دسموئیدی (DT) (15%-10%) میباشند (34-32). تومورهای دسموئیدی، نئوپلازیهای مزانشیمی با یک رشد تدریجی هستند که دارای عدم پتانسیل متاستاتیک و در عینحال رفتار تهاجمی موضعی بوده که بهدلیل رشد سریع خود و همچنین خطر عود بالا در محل، مورد توجه محققین میباشند. ریسک ابتلا به تومور دسموئیدی (DT) در بیماران مبتلا به FAP، حدود 1000 برابر بیشتر نسبت به جمعیت عادی است. اغلب تومورهای دسموئیدی در بیماران FAP، در شکم و بیشتر در نواحی دیواره شکمی یا داخل شکمی ایجاد میشوند. از عوامل ریسک ابتلا به تومورهای دسموئیدی میتوان به سابقه جراحی شکم، یک سابقه خانوادگی مثبت برای بروز دسموئید، و همانطور که گفته شد، جایگاه جهش در ژن APC، اشاره کرد. با وجود خوشخیم بودن تومورهای دسموئیدی، آنها یکی از علل اصلی مرگ در بیماران FAP هستند (35). از بدخیمیهای غیرکولون نیز میتوان به سرطان تیروئید (3%-2%)، آدنوکارسینومای مخاطی پانکراس (1%)، هپاتوبلاستوما (1%) و تومورهای مغزی (برای نمونه، بلاستومای مغزی <1%) اشاره نمود. کارسینومای تیروئیدی پاپیلاری، سومین بدخیمی رایج در ارتباط با بیماری FAP (بعد از CRC و سرطان دوازدهه) است. با اینحال، خطر ابتلا به سرطان تیروئید، پایین بوده و بین 3%-2% با نرخ تقریبی 160 برابری نسبت به جمعیت کل تخمین زده میشود (36). زنان در ابتلا به این بیماری، دارای درصد نفوذ قابلتوجهی هستند (نسبت زن به مرد 17:1) و میانگین سنی در تشخیص آنها، 27 سال میباشد. اگرچه سرطان تیروئید در بیماران FAP میتواند گرههای لنفاوی منطقهای را نیز درگیر کند و حالت چندموضعی داشته باشد، با اینحال، پیشآگاهی مناسب، در رابطه با آن بسیار مفید است (37). بلاستومای کبدی نیز یک نئوپلازی جنینی است که عمدتا در کودکان 6 ماهه تا 3 ساله رخ میدهد، اما سن تشخیص آن میتواند از مراحل قبل از تولد تا 16 سالگی باشد. اگرچه شیمیدرمانی و جراحی، برای این مورد بسیار موفقیتآمیز است، اما تخمین زده میشود که کمتر از 25% از کل بیماران، زنده میمانند. سندرم گاردنر (Gardner) نیز بهعنوان ترکیبی از درگیریهای کولورکتال و نشانههای غیرکلون، شناخته میشود، درحالیکه سندرم تورکوت (Turcot) در ارتباط با پولیپهای کولورکتال و تومورهای مغزی معرفی میگردد (38).
همبستگی ژنوتیپ - فنوتیپ: وجود طیفی از پولیپهایی که ناشی از جهش در مناطق مختلف ژن APC اند، توسط Leppert و همکاران در سال 1990 پیشنهاد شد (39). از آن پس، مطالعات متعددی، ارتباط بین نشانههای بالینی و محل قرارگیری جهشهای ژرمینال را مشخص کردند. بهطور کلی، جهشهای بین کدونهای 178 و 309 و همینطور بین کدونهای 409 و 1580 مرتبط با فنوتیپ کلاسیک FAP، واجد بیش از 100 آدنوما هستند که مربوط به اگزونهای 8-5 و 14-9 و همچنین نیمه ابتدایی اگزون بزرگ 15 میباشند (40). بیماری FAP را میتوان براساس همبستگی ژنوتیپی - فنوتیپی، به 3 دسته تقسیمبندی کرد. 1) پولیپهای تهاجمی که دارای ویژگی شروع زودتر و تعداد بیشتر پولیپها، با جهشهایی در کدونهای 1250 تا 1464 و عمدتا در کدون 1309 هستند. 2) AFAP که معمولاً با جهشهایی در انتهای ′5 (قبل از کدون 157) و انتهای ′3 (پس از کدون 1595) ژن APC و ناحیهای از اگزون 9 (کدونهای 412-213) که بهصورت متناوب تحت پیرایش قرار میگیرند، همراه هستند و در نهایت، 3) فنوتیپ حدواسط در FAP کلاسیک، مجموع جهشهایی را شامل میشود که در باقیمانده ژن APC به خصوص انتهای ′5 بین کدون 157 و 1595 به غیر از کدون 1309، واقع شده باشند (شکل 2) (41,42). همچنین جهشهای خاصی در ژن APC، بهویژه بعد از کدون 1400، مرتبط با نشانههای غیرکولون میباشند. CHRPE مرتبط با جهشهایی است که بین کدون 311 و کدون 1456 قرار گرفتهاند و حضور تومورهای دسموئیدی نیز مرتبط با جهشهای انتهای ′3 در ژن APC و بهطور کلی پاییندست کدون 1400 (2011-1445) است. حضور پولیپهای معده و دوازدهه نیز با جهشهایی در انتهای ′3 و قبل از کدون 395 و همچنین اگزون 4 و کدون 1493-564 مرتبط است (43). سایر مواردی که نشاندهنده وجود همبستگی ژنوتیپ- فنوتیپ باشد، با شواهد اندکی مشاهده شده است. در بیماران بلاستومای کبدی، تقریباً 95% از جهشها در انتهای ′5 تا ناحیه میانی ژن APC، بین کدونهای 141 و 1751 قرار گرفتهاند. تومورهای تیروئیدی با جهشهایی مابین کدونهای 140 و 1309 مرتبط هستند (شکل 2). با وجود مشاهده همبستگی ژنوتیپ- فنوتیپ، در بین افراد بیمار و حتی بین اعضای خانواده آنها، متغیرهای قابلتوجهی وجود دارند که نشاندهنده تاثیر عوامل محیطی و یا اثر ژنهای اصلاحشده دیگر است (43,44).
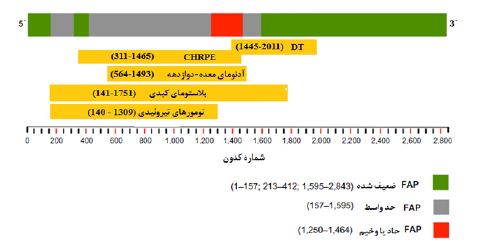
شکل 2: ارتباط ژنوتیپ-فنوتیپ در ژن APC
اختصارات: DT (تومورهای دسموئیدی)، CHRPE (هایپرتروفی مادرزادی اپیتلیوم رنگدانه شبکیه)، FAP (پولیپوز آدنوماتوز خانوادگی)
الگوریتمهای تستهای ژنتیکی: قبل از آزمایشهای ژنتیک، افراد مبتلا بهمنظور فهم جوانب مثبت و منفی آزمایشات ژنتیکی سرطان، میبایست مشاوره ژنتیکی دریافت کنند. بیماران میبایست تعیین کنند که آیا از نظر روحی برای چنین تستهایی آمادگی دارند یاخیر، و سایر عوامل (مانند محرمانه بودن اطلاعات بیمار) برای آنها توضیح داده شود. هنگامی که روند مشاوره ژنتیک بهدرستی انجام شود، در صورت مشخصشدن نوع جهش ژنی، میتوان به بستگانی که دارای ریسک ابتلا به بیماری هستند، نیز نشانههای اولیه بیماری را توضیح داد. اواسط نوجوانی، زمان مناسبی برای انجام آزمایشات ژنتیک میباشد که از نظر تشخیصی و پیشگیری از سرطان دارای اهمیت بالینی قابلتوجهی است. اگر هیچ جهش پاتوژن ژرمینالی در بیمار یافت نشد، انجام آزمایشات ژنتیکی را نمیتوان به دیگر اعضای خانواده پیشنهاد کرد و فقط انجام تستهای بالینی و مراقبتهای شخصی، برای همه بستگان درجه اول فرد، توصیه میشود. روشهای متعددی برای بررسی ژن APC مورد استفاده قرار گرفته است. تعیین توالی مستقیم ژن، از تمام 15 اگزون کدکننده ژن APC بهعنوان استاندارد طلایی در شناسایی جهشهای ژنی در نظر گرفته شده است. با اینحال، روشهای دیگری نیز استفاده میشود. در گذشته چندین آزمایشگاه، از آزمایش برش پروتئین (PTT)، بر مبنای RNA استفاده میکردند. این روش براساس تجزیه و تحلیل اندازه محصولات حاصل از رونویسی و ترجمه، در شرایط آزمایشگاهی است و حساسیت آن بین 70 تا 90 درصد است. با اینحال، روش PTT دارای معایبی از جمله: دستگاههای مورد نیاز برای آزمایش و ناتوانی در شناسایی جهشهایی که در اندازه محصول تغییری ایجاد نمیکنند، میباشد. روشهای دیگر، شامل روشهای اسکنینگ (مانند: ژل الکتروفورز کنفورماسیون رشتهای) و متعاقب آن، تعیینتوالی قطعات جهشیافته میباشد. با این وجود، هیچیک از این روشها به اندازه روش تعیین توالی مستقیم ژن، حساسیت نداشته و به همین دلیل در اکثر آزمایشگاههای بالینی، بهمنظور شناسایی جهشهای نقطهای و حذفها یا اضافهشدنهای کوچک که 85 درصد از جهشهای ژن APC را تشکیل میدهند، بهعنوان روشی استاندارد مدنظر است. 15%-10% از جهشهای باقیمانده، حذفها و مضاعفشدگیهای بزرگی هستند که میتوان از طریق روشهای تکثیر لیگاند وابسته به پروب چندتایی(MLPA) ، ساترنبلات، یا PCRکمی در زمان واقعی (Real-time quantitative PCR)، آنها را شناسایی نمود (45,46). طبق توصیه دستورالعملهای کنونی، ارزیابی FAP میبایست با استفاده از تعیینتوالی کامل ژن APC انجام شود و درصورتیکه هیچگونه جهشی یافت نشد، سپس ارزیابی بازآراییهای بزرگ کروموزومی مدنظر قرار گیرد (47).
مدیریت بالینی FAP
آدنوماهای کولورکتال و CRC: هدف از مدیریت نئوپلازی کولورکتال در بیماران FAP، پیشگیری از ابتلا به CRC میباشد. این مدیریت شامل هر دو روش جراحی و پولیپکتومی آندوسکوپی (Endoscopic polypectomy) است. در خانوادههایی با FAP کلاسیک، روش سیگموئیدوسکوپی انعطافپذیر (Flexible sigmoidoscopy)، به دلیل یک توزیع تقریبا فراگیر از آدنوماها، از جمله در ناحیه رکتوم، بهعنوان تکنیک تشخیصی مناسب درنظر گرفته میشود. سن شروع غربالگری، وابسته به میزان ریسک ابتلا به آدنوماهای بدخیم کولورکتال است. با وجود اینکه بیش از 1/5% بیماران مبتلا به FAP در بین سنین 11 تا 20 سالگی به CRC دچار میشوند، اما ریسک ابتلا به آن در بیماران کمتر از 20 سال، خیلی پایین است. بنابراین غربالگری سیگموئیدوسکوپی، میبایست از سن 12 تا 14 سالگی هر دو سال یکبار انجام شود و بهصورت مادامالعمر برای افراد ناقل جهش، ادامه یابد. پس از تشخیص آدنوماها، تا زمانی که کولکتومی مشخص شده است، کلونوسکوپی کلی نیز میبایست بهطور سالانه انجام گیرد (جدول2) (35). در موارد AFAP، از زمانی که آدنوماها در سمت راست کولون واقع میشوند، بهجای سیگموئیدوسکوپی، کلونوسکوپی توصیه میشود. در اینحالت، بهمنظور شروع تشخیص پولیپوز، از سن 18 تا 20 سالگی میبایست هر دو سال یکبار غربالگری صورت گیرد و هنگامی که آدنوماها شناسایی شوند، کلونوسکوپی میبایست بهصورت سالانه انجام پذیرد (48). بهمنظور جلوگیری از بروز و مرگ و میر ناشی از CRC پیشرفته، عمل جراحی حذفی کلون، در مرحله پیش از بدخیمی، حائز اهمیت است. در نوع کلاسیک FAP، زمانی که پولیپوز شدت مییابد، یا پولیپهای آزاردهنده (ایجاد زخم cm1 با درجه بالای دیسپلازی) شناسایی شوند، معمولاً کولکتومی بهمنظور پیشگیری از CRC توصیه میشود. اکثر بیماران مبتلا به FAP کلاسیک، بین سنین 15 تا 25 سالگی تحت عمل جراحی قرار میگیرند. راه درمانی AFAP نیز معمولاً آندوسکوپی است و در صورتی که این امر میسر نباشد، عمل جراحی بهشیوهای مشابه با FAP کلاسیک انجام میگیرد. گزینههای جراحی شامل: پروکتولکتومی با آناستوموز ایلئوآنال (IPAA) و کولکتومی توتال با آناستوموز ایلئورکتال (IRA) است. IPAA در مقایسه با IRA با کمترین میزان عوارض و معمولاً عملکرد خوب روده پس از جراحی، نسبتاً سادهتر میباشد. در مورد IPAA، جراحی گستردهتری (از جمله مداخله در لگن) موردنیاز است که منجر به کاهش باروری و بدترشدن عملکرد رودهها نیز میگردد (49). انتخاب روش جراحی، عمدتا وابسته به سن تشخیص، دسموئیدها، باروری و تعداد پولیپهای رکتال (15 تا 20 پولیپ) و همچنین تصمیم بیمار پس از دریافت اطلاعات جامع در مورد مزایا و خطرات هر کدام از روشهای درمانی میباشد (50). برخی از محققین، استفاده از شواهد مربوط به ارتباط ژنوتیپ- فنوتیپ را بهعنوان راهنما، در جراحی بیمارانی با رکتومی نسبتا نازک پیشنهاد کردهاند (43). IPAA ممکن است در بیمارانی با ژنوتیپ تشدیدیافته نیز توصیه شود، زیرا این بیماران در معرض خطر ابتلا به پولیپوز شدید راست - روده هستند که درصورت انجام IRA، به پروکتکتومی مجدد نیاز خواهند داشت. پس از جراحی، برای آندسته از بیماران دارای بقایای مقعدی، پیگیری از طریق آندوسکوپی بهدلیل وجود ریسک ابتلا به سرطان رکتال (بیش از 30% موارد) توصیه میشود. در بسیاری از مطالعات نشان داده شده است که پس از پروکتوکلکتومی (Proctocolectomy) مجدد نیز آدنوماها و گاهی اوقات حتی آدنوکارسینوماها در کیسه ایلئوس مقعد دیده شدهاند. بنابراین مراقبت بر کیسه و منطقه مقعدی که دستخوش تغییر شده است، ضرورت دارد (51). بهطور کلی، در رابطه با نشانههای غیرکلون، غربالگری میبایست در زمان شروع تشخیص پولیپها یا بین سنین 25 تا 30 سالگی صورت پذیرد. در صورت شناسایی آدنوماها، آندوسکوپی معده – دوازدهه در هر دو جهت جلویی و جانبی (به منظور رویت صحیح آمپولاواتر: Vater’s ampulla) میبایست هر پنج سال یکبار انجام گیرد. در عمل، با توجه به شیوع کم آدنوکارسینومای معدی، مراقبت از طریق آندوسکوپی دستگاه گوارش فوقاتی، بهدلیل وجود ریسک ابتلا به سرطان دوازدهه ضروری است. معده بهعنوان بخشی از این مراقبت، رویت میشود، اما بیوپسی یا پولیپکتومی (Polypectomy) تنها برای ضایعات بزرگ و غیرمعمول، بهخصوص در آنتروم، اعمال میگردد (52). به منظور استانداردسازی و مدیریت پولیپهای دوازدهه در بیماران FAP، Spigelman و همکارانش، یک سیستم طبقهبندی را براساس چهار متغیر پیشآگاهی: تعداد پولیپها، اندازه آنها، بافتشناسی و درجه دیسپلازی معرفی کردند (جدول 3) (53). در مرحله I (با 4 امتیاز) حدمتوسطی از بیماری مشاهده میشود و در مراحل III و IV (با امتیاز بالاتر از 6) پولیپهای وخیمی در دوازدهه، با ریسک قابلتوجهی برای ابتلا به سرطان دوازدهه (7 تا 36 درصد)، بروز مییابند. تقریبا 80% بیماران، در مرحله I تا III و 10 تا 20 درصد موارد، در مرحله IV بیماری هستند. شواهد موجود حاکی از آن است که معاینه دوازدهه، از طریق کرومواندوسکوپی (Chromoendoscopy) یا تصویربرداری موجب افزایش تشخیص آدنوماهای دوازدهه میگردد، اما منجر به تغییر قابلملاحظهای در مراحل Spigelman نمیشود. مدیریت بیمارانی با چندین آدنومای بزرگتر (مرحله III یا بالاتر) ، چالشبرانگیز بوده و میبایست در مراکز بالینی اختصاصی انجام شود (54). میزان عود پیشرفت آدنوما، بعد از درمان آندوسکوپی نیز بسیار زیاد است (بیش از 50%) و درمان این موارد نیز با عوارض خطرناکی همچون خطر سوراخشدن، خونریزی و آسیب پانکراتیک همراه است (52(. از آنجاییکه حذف تمام آدنوماها امکانپذیر نیست، اولویت اول، حذف آدنوماهای بزرگ (بیش از cm1) یا آدنوماهایی با درجه دیسپلازی بالا، با هدف به تاخیرانداختن یا اجتناب از عمل جراحی، میباشد. در بیمارانی با درجه IV وخامت، جراحی غالبا ضروری است که شامل: دئودنوتومی (Duodenotomy) با پولیپکتومی، دئودنکتومی پانکراس و پانکراتکتومی دوازدهه میباشد (55).
جدول 2: توصیههای مدیریت بالینی برای FAP
.JPG)
اختصارات: پولیپوز آدنوماتوز خانوادگی:FAP، توموگرافی رایانهای : CT، تصویربرداری رزونانس مغناطیسی : MRI. پولیپهای معده- دوازدهه
جدول3: رده بندی بر اساس معیارهای Spigelman
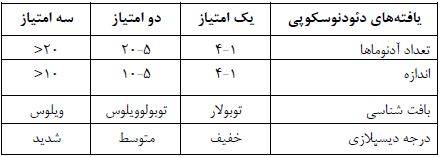
مرحله بندی بر اساس امتیاز: مرحله 0: 0 امتیاز، مرحله I: 4 امتیاز، مرحله II: 6-5 امتیاز، مرحله III: 8-7 امتیاز، مرحله IV: 12-9 امتیاز.
مدیریت بالینی سایر تومورها: با توجه به افزایش خطر ابتلا به سرطان تیروئید، متخصصین بر این باورند که بررسی گردن و یا تیروئید، باید از سن 30-25 سالگی و بهصورت سالانه انجام شود. پیشرفت تومور دیسموئید (DT) نیز عمدتا وابسته به یک سابقه خانوادگی مثبت، جراحی شکم، و محل بروز جهش بوده و میتواند در داخل یا دیواره شکمی ایجاد شود. DT را میتوان از طریق توموگرافی رایانهای (CT) یا تصویربرداری رزونانس مغناطیسی (MRI) تشخیص داد. گزینههای درمانی آن عبارتند از: درمان دارویی (داروهای ضدالتهاب غیراستروئیدی NSAID و یا آنتیاستروژنها)، شیمیدرمانی، مداخله جراحی و یا پرتودرمانی (56). طبق شواهد بالینی، اثرگذاری این درمانها نسبتاً ضعیف بوده و مبتنی بر مطالعات محدودی است. بااینحال، در صورت عدم بروز عوارض و به دلیل بالابودن میزان عود DT، هرگونه مداخله از طریق عمل جراحی تومورهای داخل شکمی، میبایست به تعویق بیفتد. متخصصین در بیمارانی با DTهای بزرگ یا در حال رشد، داروهای تاموکسیفن با سولینداک (Sulindac) را بهعنوان خط اول درمان توصیه میکنند و هنگامیکه بیمارانی با تومورهای داخل شکمی به این درمان پاسخ نمیدهند، شیمیدرمانی یا پرتودرمانی را تجویز میکنند. تومورهای دسموئیدی واقع در دیواره شکمی و DTهای بافت مزانتریک، میبایست بهطرز متفاوتی از هم، مدنظر قرار گیرند. عمل جراحی معمولاً بهعنوان خط اول درمان، برای مداوای DTهای دیواره شکمی مورد استفاده قرار میگیرد. با اینحال، در مورد DT مزانتریک با توجه به شدت بیماری، عوارض احتمالی و سنجیدن مزیتهای روش درمان، نسبت به ریسک روشهای دیگر، استراتژی بهینه بهصورت شخصی انتخاب میشود (57). سایر بدخیمیهای غیر رودهای (پانکراتیک، مغزی و آدرنال) دارای شیوع بسیار کمتری میباشند که استفاده از مطالعات پرهزینه دیگری برای آنها توصیه نمیشود. با اینحال، تستهای نظارتی در بیمارانی، با یک سابقه خانوادگی قوی، در رابطه با هریک از این علایم خاص غیر رودهای و آن دسته از افرادیکه دارای نشانههای منتسب به این بیماریاند، میبایست انجام شود.
روشهای پیشگیری شیمیایی: در بیماران FAP بهمنظور پیشگیری، از روشهای شیمیایی، نظیر استفاده از داروهای NSAID نیز استفاده میشود. NSDAIها با عملکرد آنزیم COX-1 و COX-2 (Cyclooxygenase) در تداخل هستند و از تبدیل اسید آراشیدونیک به پروستاگلاندین¬ها که در پیدایش درد نقش دارند، جلوگیری می¬کنند (58). سولینداک نیز بهعنوان یکی از اولین داروهایی است که در این بیماری موثر بوده است. استفاده طولانیمدت از این دارو، منجر به کاهش 50 درصدی آدنوماهای کولورکتال در کولون و همچنین در رکتوم بیماران پس از کولکتومی میشود، اما بر پولیپوز دوازدهه موثر نیست. با اینوجود، داروی سالینداک از پیشرفت آدنوماها در بیماری FAP جلوگیری نمیکند (59). داروی سلکوکسیب (Celecoxib) نیز بهعنوان مهارکننده انتخابی پروتئین (cyclooxygenase-2) COX-2، همراه با عوارض گوارشی کمتر نسبت به داروی سالینداک، و بهمنظور کاهش 28 درصدی تعداد آدنوماهای کولورکتال معرفی شده است و توانسته است تعداد آدنوماهای دوازدهه را در بیماران، کاهش دهد. با اینوجود، در مصرفکنندگانی که بهصورت طولانیمدت از مهارکننده انتخابی COX-2 دیگری (مانند روفکوکسیب: Rofecoxib) استفاده میکردند، عوارض قلبی- عروقی (انفارکتوس یا سکته قلبی) گزارش شده است و بنابراین نقش و اثر این داروها، همچنان مورد بحث باقی میماند و میبایست تنها در بیماران انتخابشدهای که بدون فاکتورهای خطر و ریسک قلبی - عروقی هستند، مدنظر قرار گیرند (60). با این وجود، NSAIDها (سولینداک و سلکوکسیب) را نمیتوان جایگزین عمل جراحی در بیماران مبتلا به FAP کولون نمود و احتمالاً نقش آنها در به تعویق انداختن عمل جراحی، در مبتلایان به پولیپوز کولون یا بیمارانی با پولیپوز رکتال پس از کولکتومی میباشد. در رابطه با پولیپوز دوازدهه، بهاین دلیل که گزینههای درمانی، اعم از اندوسکوپی و جراحی در برخی موارد همراه با عوارض چشمگیری هستند، مصرف داروی سلکوکسیب برای بیماران مبتلا به پولیپوز شدید دوازدهه (مرحله III یا IV)، امری قابل قبول میباشد. اگرچه داروی سلکوکسیب برای درمان بیماری FAP در چندین کشور مورد تایید واقع شده است، اما برخی متخصصین به دلیل اثرات و عوارض قلبی - عروقی، نسبت به تجویز آن بیرغبت بوده و در نتیجه این دارو بهندرت مورد استفاده قرار میگیرد (61).
:MAP MAP یک اختلال ارثی با الگوی اتوزومال مغلوب است که به علت جهشهای ژرمینال دوآللی در ژن MutYH ایجاد میشود. این بیماری برای اولینبار در سال 2002 توسط AL-Tassan و همکارانش، در یک خانواده بریتانیایی با سه عضو مبتلا و توارث مغلوب آدنوماهای متعدد کولورکتال و کارسینوما، مورد بررسی قرار گرفت. علایم بالینی در بیماران مبتلا به MAP، دارای تنوع زیادی میباشد، اما معمولاً بهصورت یک فنوتیپ پولیپوز تضعیفشده، همراه با تعداد کمتر از 100 آدنوما بروز مییابد. برخی از بیماران، به علایمی غیر از روده نیز دچار میشوند که در بیماران FAP غیرقابل تشخیص است (62).
ژن MutYH: ژن MutYH بر روی کروموزوم 1p34.1 واقع شده و دارای 16 اگزون کدکننده یک پروتئین 535 آمینواسیدی میباشد (شکل 1B). این ژن یک عضو از سیستم ترمیمی برش بازی (BER) را کد میکند. این سیستم، متشکل از 3 آنزیم (MutYH, OGG1, MTH1) میباشد که در محافظت سلولها در برابر اثرات جهشزای متابولیسم هوازی و بهویژه اکسیداسیون نوکلئوتید گوانین که منجر به تشکیل 8-اکسوگوانین (oxoG-8: (8-hydroxyguanine, 8-oxo-Gua, or OH8Gua میگردد، شرکت میکند. پروتئین MutYH همرا با OGG1 و MTH1 از بروز جهشهای سوماتیک که ناشی از 8-اکسوگوانین بوده و تمایل زیادی به جایگزینی نوکلئوتید آدنین (A) به جای سیتوزین دارند، ممانعت بهعمل میآورد. بهویژه آنکه پروتئین MutYH مسئول حذف آدنینهایی است که به اشتباه با 8-اکسوگوانین جفت شدهاند (63). در غیاب یک کپی عملکردی از پروتئین MutYH به دلیل جهشهای دو آللی، زمانی که یک 8-اکسوگوانین، بهصورت جفت باز اشتباه در الگوی DNA قرار گیرد، در دور بعدی همانندسازی جفت بازG:C به T:A تبدیل میشود. به همین علت، جهشهای سوماتیکG:C به T:A در ژنهایی مانند: APC یا KRAS اغلب در آدنوماها و تومورهای مرتبط با MutYH رخ میدهد. بهعنوان مثال، در ژن KRAS (جهش c.34G>T در کدون 12) اغلب در بیماران MAP مبتلا به CRC بیشتر است (64%). بنابراین آنالیز جهشهای سوماتیک در ژن KRAS بهعنوان یک تست پیشغربالگری برای شناسایی بیماران CRC که واجد شرایط برای آزمایشات ژنتیکی در مورد جهشهای ژرمینال ژن MutYH هستند، توصیه شده است (64). از آنجاکه در بیماران مبتلا به MAP آدنوماهای معمولی و همچنین پولیپهای دندانهای (پولیپهای هایپرپلازی) میتوانند حضور داشته باشند، وجود دو مسیر مجزا پیشنهاد شده است که یکی از آنها، بروز جهشهایی در ژن KRAS و یا ژن APC است که منجر به آدنوماهای معمولی شده و مسیر بعدی، بدون رخداد جهش در ژن APC و فقط از طریق جهشهایی در ژن KRAS است که منجر به پولیپهای هایپرپلازی میشود (65).
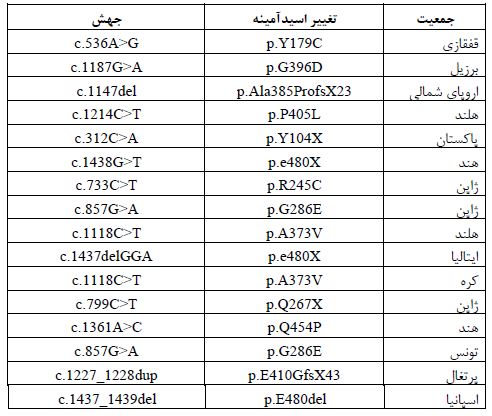
نگاهی به جهشهای ژن MutYH: تاکنون بیش از 300 واریانت منحصر بهفرد و قریب به 80 جهش بیماریزا در این ژن شناسایی شده است. اکثر آنها از نوع جایگزینیهای بدمعنی و تعداد کمی از جهشها نیز از نوع کوتاهکننده یا جایگاه پیرایش هستند. اگرچه در موارد نادری، جهشهایی از نوع حذفهای ژنومی بزرگ، تغییر چارچوب و جهشهای بیمعنی نیز گزارش شده است. جهشها تقریبا در تمامی اگزونها (به غیر از اگزون 1 و 2) گزارش شده است. غالبترین جهشهای بدمعنی، دو جهش واقع در نقاط داغ ژن میباشند که شامل: p.Y179C (c.536A>G; p.Tyr179Cys) در اگزون 7 و جهش p.G396D (c.1187G>A; p.Gly396Asp) در اگزون 13 هستند و حدود 70 تا 80 درصد از تمام جهشهای جمعیتهای اروپایی را در بر میگیرند (جدول 4) (66). مطالعات گذشته، اختلافات جغرافیایی و نژادی را در مورد فراوانی جهشهای ژن MutYH را بررسی نمودهاند. همانطور که ذکر شد، دو جهش p.Y179C و p.G396D در جمعیتهای اروپایی، بهمراتب شایعترین واریانتهای بیماریزا میباشند. علاوه براین، با توجه به تفاوتهای قومی و جغرافیایی، واریانتهای مختلف، دارای نقش اثرگذاری در سایر جمعیتها هستند، بهعنوان مثال: جهش بیمعنی p.Y104X در بیماران پاکستانی و جهش بیمعنی p.E480X در بیماران هندی بسیار حائز اهمیتاند (67). جهشهای خاص دیگری نیز در جمعیتهای ژاپنی و جنوب اروپا گزارش شده است (جدول 4). مطالعات متعددی به بررسی فراوانی دو نوع متداول از جهشهای بدمعنی (p.Y179C و p.G396D) پرداختهاند و پیش بینی شده است که تقریبا 1 تا 2 درصد از جمعیت عمومی (با منشا اروپایی) ناقل این جهشها باشند (68).
جدول 4: جهشهای شایع ژرمینال در ژن MutYH در جمعیتهای مختلف
فنوتیپ: طیف بالینی جهشهای ژرمینال در ژن MutYH هتروژنوس بوده و میتواند گستره وسیعی از فنوتیپها را شامل شود.
پولیپوز آدنوماتوی کلاسیک و تضعیفشده: اکثر ناقلین جهش دو آللی در ژن MutYH دارای ده تا چند صد پولیپ هستند و در تعداد بسیار کمی از بیماران بیش از 500 پولیپ نیز گسترش یافتهاست. در ابتدا، جهشهای دو آللی در یک سوم از بیماران AFAP با APC منفی، همراه با بیش از 15 آدنوما و در حدود 10 درصد بیماران مبتلا به FAP کلاسیک با APC منفی و بهویژه، در آن دسته از مواردی که دارای یک الگوی توارثی مغلوب بودند، شناسایی شد (69). طی مطالعه دیگری نیز در مورد جهشهای دو آللی در ژن MutYH، مشخص گردید که این جهشها در 95 از 119 بیمار با بیش از 1000 آدنوما و 94 از 1338 بیمار با 100 تا 999 آدنوما و 233 از 3253 بیمار با 20 تا 99 آدنوما و 37 از 970 بیمار با 10 تا 19 آدنوما مشاهده شد (70). سندرم پولیپوز پایهدار و بیماران مبتلا به پولیپهای چندگانه آدنوما و پولیپهای پایهدار، بهطور شایع در MAP یافت میشوند. در تعداد کمی از بیماران MAP، بهمنظور بررسی پولیپوز آدنوماتوز، از معیارهای WHO برای سندرم پولیپوز پایهدار استفاده میشود (زمانی که یک بیمار حداقل دارای یکی از معیارهای زیر باشد: 1- وجود حداقل پنج پولیپ پایهدار در نزدیکی کولون سیگموئید که دو تا از آنها دارای قطری بیش از mm 10 باشند، 2- هریک از پولیپهای پایهداری که نزدیک کولون سیگموئید فردی با پولیپوز پایهدار درجه اول ایجاد شود 3- بیش از 20 پولیپ پایهدار که در اندازههای متفاوت در سرتاسر کولون توزیع شده باشند) (71). در مطالعهای که بر روی بیماران شناسایی شده با حداقل ده پولیپ از هر نوع بافتی (شامل پولیپهای آدنوماتوز و پایهدار) متمرکز شده بود، 6/7% (405/27) از بیماران، بهصورت ناقلین جهش دو آللی در ژن MutYH مشاهده شدند (71).
CRC بدون پولیپوز: در مطالعات CRC مبتنی بر جمعیت که در آن، بیماران بر اساس تشخیص CRC مورد بررسی قرار گرفته بودند، جهشهای دو آللی در ژن MutYH در 2 تا 3 درصد جمعیت یافت شد. در چندین مطالعه دیگر مبتنی بر جمعیت، بیش از یک سوم ناقلین جهش دو آللی در ژن MutYH، دارای فنوتیپ پولیپوز نبودند. بر ایناساس، جهشهای دو آللی در ژن MutYH همیشه با یک فنوتیپ پولیپوز همراه نیستند، بنابراین، این گونه جهشها میبایست در بیماران مبتلا به CRC زودهنگام (قبل از سن 50 سالگی) در نظر گرفته شوند، بهویژه اگر احتمال مبتلا بودن به سندرم لینچ در بیماران، رد شده باشد. بااینحال، فنوتیپهای غیر معمول نیز گزارش شده است (72). اخیرا در یک مطالعه، ژن MutYH در 85 مورد مشکوک به سندرم لینچ، همراه با تومورهایی مشاهده شدند که نشاندهنده نقص سیستم ترمیمی جفت باز اشتباه و همچنین بدون هیچ جهش ژرمینال APC قابل تشخیص بودند، این بیماران ناقل جهش دوآللی (p.Y179C) همراه با CRC گسترشیافته، کارسینومای اوروتلیال و کارسینومای غدد چربی، شناسایی شدند. در این بیماران خاص، تعیینتوالی تومور، دو جهش سوماتیکی در سیستم ترمیم جفت باز را نشان داد که به اینترتیب فنوتیپ بیماری قابل توضیح بود (73).
جهشهای هتروژنوس در ژن MutYH و خطر ابتلا به CRC: ریسک ابتلا به CRC در افراد دارای جهشهای یکآللی (هتروزیگوت) بهعنوان مبحثی مهم و چالشبرانگیز بوده است. چندین متاآنالیز، همراه با OR (نسبت احتمالات) یا RR (نسبت نسبی) بین بیماران و افراد عادی جمعیتهای مختلف انجام شد که بیشتر آنها هیچ گونه تفاوت آماری را نشان ندادند. اخیراً طبق جدیدترین یافتهها، مشخص شده است که فراوانی ناقلین جهشهای یکآللی که دارای یک سابقه خانوادگی از بیماری بودند، نسبت به افراد کنترل (P=0.02) بیشتر بوده و پیشنهاد میشود که ناقلین جهشهای یکآللی با سابقه خانوادگی CRC، نیز ممکن است در معرض خطر بیشتری باشند (74,75).
علایم و نشانههای غیرکولون: از آنجاییکه استرس اکسیداتیو در بافتهای مختلف وجود دارد، میتوان انتظار داشت که یک ژن MutYH معیوب، باعث ایجاد نئوپلازیهایی در سایر اعضای بدن نیز گردد. Vogt و همکارانش اخیرا طیفی از خصوصیات غیرکولون گروه بزرگی از بیماران MAP را (276 بیمار مبتلا به MAP) تعیین نمودند. در این مطالعه 17% بیماران، در معرض خطر ابتلا به پولیپوز دوازدهه در طول مدت عمر خود بوده و 4% ریسک ابتلا به سرطان دوازدهه و 38% در معرض خطر ابتلا به هر یک از سرطانهای غیر از روده میباشند. بروز بدخیمیهای غیر رودهای، در بین بیماران تقریبا دو برابر بیشتر از جمعیت کلی بوده و افزایش قابلتوجهی در بروز سرطانهای تخمدان، مثانه و پوست، و روندی افزایشی در ریسک ابتلا به سرطان پستان برای آنها وجود دارد (76). سن متوسط در تشخیص سرطانهایی غیر از روده، بین سنین 51 تا 61 سال متغیر است. سایر عوارض دیده شده در بیماران FAP اعم از FGP معده، لیپوما، CHRPE، کیست اپیدرموئید، DTو کارسینومای تیروئیدی، در شمار اندکی از بیماران MAP نیز گزارش شده است. بهطورکلی، میزان بروز علایم مرتبط با FAP، در بیماران MAP با نسبت کمتری دیده میشود (77).
همبستگی ژنوتیپ- فنوتیپ: همبستگی ژنوتیپ- فنوتیپ، همانند ژن APC در جهشهای ژن MutYH نیز توصیف شده است. برخی مطالعات نشان دادهاند که طبق بررسیهای ژنتیکی، جهش p.Y179C نسبت به جهش نقطهای G396D، منجر به کاهش هرچه بیشتر فعالیت گلیکولازی پروتئین MutYH میگردد و در بیمارانی با جهشهای دو آللی Y179C نسبت به جهشهای G396D، با یک فنوتیپ شدیدتری (بهعنوان مثال،شروع زودتر و تعداد بیشتر پولیپها) همراه است (71). در مطالعه دیگری نیز، واریانت p.G396D همراه با پیشرفت پولیپهای پایهدار در بیماران MAP گزارش شده است (78).
الگوریتمهای تستهای ژنتیکی: بیمارانی که دارای یک شکل تضعیف شدهای از پولیپوز آدنوماتوز یا FAP کلاسیک با یک الگوی توارثی مغلوب هستند، مشکوک به جهشهای دو آللی در ژن MutYH میباشند. همچنین در بیماران مبتلا به CRC تا قبل از سن 50 سالگی و در بیماران دارای پولیپهای متعدد کولون (بیشتر از 10 عدد، از جمله آدنوماتوز و پایهدار) باید بیشتر مدنظر قرار گیرند. هنگام یافتن یک فرد ناقل جهشهای دو آللی در ژن MutYH، آزمایشات ژنتیکی را میتوان برای بستگان درجه اول او نیز پیشنهاد داد، بهویژه خواهر و برادرهایی که به میزان 25 درصد دارای ریسک ناقلبودن جهشهای دو آللی هستند. در بیماران MAP احتمال ریسک ابتلا به این بیماری در فرزندان آنها، بستگی به وضعیت ژنتیکی همسر آن فرد بیمار دارد. آنالیز جهشهای ژرمینال، معمولا شامل دو نوع از متداولترین جهشها (p.G396D و p.Y179C) میباشد. تعیینتوالی کامل ژن در موارد زیر توصیه میشود: 1) افرادی که ناقل یک یا دو جهش متداول هستند، 2) افرادی که از نظر نژادی هندی و اروپایی نیستند و 3) در افرادی با نژاد هندی و اروپایی با سابقهی خانوادگی که برای جهشهای شایع، منفی بوده باشند. از آنجا که حذفها و مضاعفشدگیهای بزرگ، بهطور فوقالعادهای در ژن MutYH گزارش شدهاند، دیگر نیازی به انجام روشهایی برای آنالیز این جهشها نیست (63).
مدیریت بالینی: پروتکل مراقبتی پیشنهادشده برای هر دو حالت بیماران MAP و AFAP مشابه میباشد. افراد میبایست از سن 18 تا 20 سالگی کولونوسکوپی را شروع کرده و هر دو سال یکبار و بهصورت مادامالعمر آن را ادامه دهند. مدیریت پولیپهای کولورکتال، شبیه آن چیزی است که برای بیماران AFAP گزارش شده است. در برخی از بیماران، به دلیل فنوتیپ تضعیفشده، احتمال از بینرفتن این پولیپها در طی آندوسکوپی وجود دارد. با اینحال، در صورت نیاز به جراحی، تصمیماتی مشابه با AFAP اتخاذ میشود. در بیماران مبتلا به MAP، آندوسکوپی نواحی فوقانی، همانند آنچه که برای بیماران AFAP بیان شده است، در سنین 25 تا 30 سالگی توصیه میشود. در این شرایط، هیچگونه شواهدی مبنی بر مفیدبودن روشهای پیشگیری با داروهای شیمیایی وجود ندارد (62).
نتیجهگیری
FAP یک سندرم اتوزومال غالب است که مسئول تقریباً 1٪ موارد CRC محسوب میشود. یک بیمار مبتلا به FAP احتمال بسیار بالایی برای ابتلا به CRC دارد، اما ممکن است سایر تظاهرات و بدخیمیهای خارج کولونی را نیز بروز دهد. این امر، اهمیت تشخیص زودهنگام و دقیق و نظارت فعال بیماری را نشان میدهد، بنابراین بهبود بیشتر ابزارهای تشخیصی را ضروری میکند. مطالعات متعددی بهطور خاص برای تهیه پانلهای چندژنی در تشخیص FAP انجام شده است و تلاش برای شناسایی واریانتهای ژنی که در حالحاضر ناشناخته ماندهاند، ادامه دارد. FAP غالباً در اثر جهشهای نوکلئوتیدی در ژن APC (کروموزوم 5q21) ایجاد میشود. یک شکل تضعیفشده از FAP نیز وجود دارد که با ایجاد کمتر از 100 آدنومای کولورکتال و شروع تاخیری CRC مشخص میشود. پولیپوز وابسته به MutYH (MAP)، الگوی اتوزومال مغلوب دارد و ناشی از جهشهای نوکلئوتیدی در ژن MutYH است. نظارت دقیق آندوسکوپی برای همه بیماران FAP، MAP و اعضای خانواده در معرض خطر آنها، توصیه میشود. علاوه بر این، تحقیقات بیشتر در تعیین اهمیت تظاهرات و بدخیمیهای خارج کولونی FAP و ارتباط آن با سایر تظاهرات بیماری، ممکن است اطلاعاتی را فراهم کند که بر استراتژیهای مراقبتی و درمانی، در آینده تأثیرگذار باشد. در نهایت، هدف همه این تحقیقات، کاهش عوارض بیماری و تلاش جهت افزایش طول عمر و کیفیت زندگی قابل قبول برای افراد آسیبدیده است.
سپاسگزاری
نویسندگان مقاله از حمایتهای دانشگاه یزد در انجام این پژوهش تشکر و قدرانی میکنند.
حامی مالی: ندارد.
تعارض در منافع: وجود ندارد.
مشارکت نویسندگان
محمد مهدی حیدری در ارائه ایده، مهری خاتمی در طراحی مطالعه، الهام افخمی عقدا و مریم طهماسبی در جمعآوری دادهها، زهرا شاکر اردکانی در تجزیه و تحلیل دادهها مشارکت داشته و همه نویسندگان در تدوین، ویرایش اولیه و نهایی مقاله و پاسخگویی به سوالات مرتبط با مقاله سهیم هستند.
پولیپوز آدنوماتوز خانوادگی (FAP) یک سندرم ارثی است که از طریق پیشرفت آدنوماهای متعدد در کولورکتوم و افزایش ریسک ابتلا به سرطان کولورکتال (CRC) و همچنین وجود علایمی در بافتهایی غیر از بافت کولون تظاهر مییابد. جهشهای ژرمینال ژن پولیپوز آدنوماتوز کولی (APC) در ابتدا در سال 1911 بهعنوان عامل بیماری FAP و با الگوی وراثتی اتوزومال غالب توصیف شد (1,2). از آن بهبعد، شواهد متعددی از جمله: یافتههای پاتوفیزیولوژی، ژنتیکی، فنوتیپیکی و علایم بالینی منجر به ابداع روشهای پیشگیرانه مناسبی در بیماران شده است. در سال 2002، ژن MutYH، بهعنوان یکی دیگر از ژنهای دخیل در پولیپوز شناسایی شد و مشخص گردید که جهشهای دوآللی آن، منجر به ایجاد FAP، با الگوی وراثتی اتوزومال مغلوب میشود که معمولا از آن بهعنوان پولیپوز وابسته به MutYH، (MAP) یاد میگردد (3). با اینحال، 20 تا 35% موارد نوظهور (Denovo) FAP، بدون شواهد بالینی یا ژنتیکی در والدین بیمار گزارش میشوند. به همین دلیل، با توجه به افزایش خطر بدخیمی، پروتکلهای غربالگری ژنتیکی برای بیماران مبتلا به FAP پیشنهاد شده است. نتایج تحقیقات قبلی ما در بیماران FAP نیز نقش پاتوژنز جهشهای ژنهای میتوکندری، علاوه بر ناهنجاریهای ژنوم هستهای، را در بیماران خانوادگی و تکگیر تایید کرد (7-4). با اینحال، در مقاله حاضر، تمرکز بر تغییرات ژنتیکی در ژنوم هستهای دخیل در فرایند بیماریزایی FAP است.
FAP وابسته به APC (APC-FAP): FAPوابسته به APC (OMIM#175100) یک بیماری ارثی با الگوی اتوزومال غالب است که از طریق گسترش چندین آدنوما در سرتاسر کولورکتوم، تظاهر مییابد. کمتر از 1% از تمام سرطانهای کولون ( (CRCرا این نوع FAP تشکیل میدهد که شیوع آن یک در هر 7000 تا 10000 نفر بوده و شایعترین پولیپوز دستگاه گوارش محسوب میشود (2).
ژن APC : APC یک ژن سرکوبگر توموری است که در موقعیت کروموزومی 5q21-22 قرار گرفته است. این ژن دارای 15 اگزون است که اگزون 15 آن، به تنهایی 75% از توالی کدکننده را شامل می¬شود و بهعنوان اصلیترین اگزون مورد هدف جهشهای ژرمینال و سوماتیک به حساب میآید. ژن APC کدکننده پروتئینی با 2843 آمینواسید (kDa310) میباشد که در مسیر سیگنالینگ Wnt بهطور مستقیم نقش دارد (8). این پروتئین چندعملکردی، حاوی چندین ایزوفرم، موتیف و دامینهای آمینواسیدی در سلول میباشد که به آن اجازه میدهد تا با مولکولهای متعدد دیگر تعامل داشته باشد (شکل 1). پروتئین APC بهعنوان یک سرکوبکننده توموری، خود از طریق تنظیم منفی انکوپروتئین-catenin β تنظیم میشود. پروتئین APC منجر به یوبیکوییتینهشدن(Ubiquitination) و تکهتکه شدن catenin –β میگردد، بنابراین در غیاب آن،catenin –β در هسته تجمع مییابد و با فاکتورهایی برهم کنش میکند که در تنظیم نسخهبرداری ژنهای درگیر در سیکل سلولی، تکثیر، تمایز، مهاجرت و آپوپتوز نقش دارند (9). علاوه بر این، APC از طریق تثبیت میکروتوبولها، منجر به افزایش پایداری کروموزومها نیز میگردد، در نتیجه غیرفعال ساختنAPC موجب تفرق ناصحیح یا عدم تفرق صحیح کروموزومها و نقص در فرآیند میتوز می¬شود (10). در اکثر موارد، FAP به دلیل جهشهای ژرمینال در ژن APC ، ایجاد میشود. افرادی با یک جهش ژرمینال در ژن APC دارای چندین آدنوما در ناحیه کولورکتوم هستند که این علایم، به دلیل غیر فعالسازی آلل سالم در اثر بروز جهشهای سوماتیک در ژن APC یا از دستدادن حالت هتروزیگوسیتی (LOH) در این لوکوس ایجاد میشوند (11). اخیرا در یک مطالعه تجربی، در 80% از افراد مبتلایی که دارای بیش از 1000 آدنوما بودند، در بیش از نیمی از افرادیکه دارای 100 تا 999 آدنوما بودند، در 10% از افراد مبتلا با 20 تا 99 آدنوما و در 5% از بیماران با 10 تا 19 آدنوما، جهش در ژن APC مشاهده گردید (12). هر چند که FAP نوعی بیماری با نحوه توارث اتوزومال غالب است، اما بیش از 25% از مبتلایان، دارای جهشهای ژرمینال از نوع نوظهور (denovo) هستند (13). از زمان شناسایی ژن APC، بیش از 1100 جهش ژرمینال بیماریزا (پاتوژن) در این ژن گزارش شده است. اکثر این جهشها از نوع تغییرات نوکلئوتیدی کوتاهکننده (Truncating) هستند که شامل: جهشهای بیمعنی (28%)، اضافهشدنهای کوچک نوکلئوتیدی (10%) یا حذفهای کوچک (46%) بوده و منجر به تولید پروتئین ناقص میشوند (14,15). همچنین جهشهای بدمعنی (3%) و تغییرات بزرگی (مانند: حذفهای تک اگزونی یا چند اگزونی و مضاعفشدگیهای نوکلئوتیدی) (13%) نیز با وجود اینکه نادر هستند، اما در بیماران گزارش شدهاند (16). در ذیل به چند مورد از چنین جهشهای مهمی در ژن APC اشاره میشود:
1) جهشهای کوتاهکننده: همانطور که ذکر شد، اکثر جهشهای ژن APC که مرتبط با بیماری FAP هستند، منجر به تولید پروتئین ناقصی میشوند که عمدتا ناشی از تغییر در چارچوب خواندنی یا یک جهش بیمعنی میباشد. جهشهای C>T، از متداولترین جهشهای بیمعنی در این ژن هستند. مشخص شده است که اکثر جهشهای ژرمینال در ژن APC در ناحیه ′5 ژن رخ میدهند که منجر به حذف اکثر تکرارهای اسیدآمینهای میشوند که در تنظیم ژنی در سطح β- کاتنین، و تکرارهای SAMP که در باندینگ آکسین درگیرند، نقش دارند (شکل A1) (17,18).
2) جهشهای بدمعنی: در تحقیقات گذشته، بیش از 60 جهش بدمعنی مختلف، که همگی واریانت ژنی محسوب شده و با وجود پایینبودن میزان شیوع، بهعنوان جهشهایی که بهصورت بالقوه بیماریزا هستند، توصیف شدهاند. Ile1307Lys و Glu1317Gln دو نوع از فراوانترین واریانتهای بدمعنی اند که تاکنون در این ژن گزارش شدهاند. هرچندکه واریانت Ile1307Lys که در 6% از یهودیان اشکنازی وجود دارد، منجر به فنوتیپ پولیپوز نمیشود، اما موجب افزایش 10 تا 20 درصدی ریسک ابتلا به CRC میگردد. واریانت Glu1317Gln نیز با یک ریسک متوسط برای خطر آدنوما و CRC همراه است (19).
3) جهشهای پیرایشگر (Splicing) و تغییرات بزرگ (Gross): این نوع جهشها موجب تغییر در الگوی پیرایش ژن و حذفشدگیها یا مضاعفشدگیهای نوکلئوتیدی میشوند. دادههای اخیر نیز حاکی از این موضوع هستند که بهویژه تغییرات ژنی بزرگ، بر روی پروموتور نواحی کدینگ موثر بوده و در بیش از 20% از خانوادههای FAP گزارش شده است (20).
4) جهشهای نوظهور و موزاییسم رده سلولهای زاینده: بسیاری از جهشهای ژرملاین در ژن APC، به ارث میرسند، اما میتوانند در فرد بیماری حتی بدون سوابق خانوادگی نیز به صورت جهشهای نوظهور، رخ بدهد که در بین 11% تا 25% از کل مبتلایان به FAP دیده میشوند. نرخ برآورد جهشهای نوظهور، بین 106× 9-4 جهش/گامت/ نسل میباشد و میزان جهشزایی آن طی اووژنز و اسپرماتوژنز برابر است. درصد قابلتوجهی از جهشهای نوظهور، به شکل موزاییک بهوجود میآیند که تنها بر روی یک رده از سلولهای فرد مبتلا تاثیر میگذارند (طبق برآوردها، یک پنجم از جهشهای نوظهور در بیماری FAP از نوع موزاییک هستند) (21).
5) نقاط داغ (Hotspots) جهش: نقاط داغ جهش در ژن APC در قسمت ′5 اگزون 15 و در کدونهای 1309 و 1061 واقع شدهاند که تقریبا 11% تا 17% از تمام جهشهای ژرمینال را تشکیل میدهند. همچنین، بهدلیل تجمع جهشها از کدون 1250 تا 1464، از این ناحیه بهعنوان ناحیه خوشهای جهش (MCR) یاد میکنند (شکل 1) (22). نوع جهش ژرمینال در ژن APC، ماهیت ضربه دوم را برای این ژن مشخص میکند. در صورتیکه جهش ژرمینال، مابین کدونهای 1194 و 1392 اتفاق بیفتد، به احتمال خیلی قوی، ضربه دوم بهصورت فقدان آللی در این ژن بروز مییابد. درمقابل، اگر جهش ژرمینال در خارج از این ناحیه رخ دهد، ضربه دوم به احتمال زیاد، یک جهش ناقصکننده در MCR است (23).
فنوتیپها: FAP کلاسیک و FAP تضعیفشده (AFAP): مطابق با تعداد پولیپها و سن فرد، دو نوع فنوتیپ اصلی برای بیماری FAP بیان میشود (جدول 1):
FAP کلاسیک: از طریق وجود صدها تا هزاران پولیپ آدنوماتوز در سرتاسر کولون و رکتوم مشخص میشود. معمولاً در زمان نوجوانی، پولیپها در رکتوسیگموئید در اندازههای کوچک، شناسایی میشوند و پس از آن اندازه و تعداد آنها افزایش مییابد. آدنوماها تقریبا در نیمی از بیماران از 15 سالگی و در 95% از بیماران در سن 35 سالگی رشد میکنند. کاملا بدیهی است که سن ابتلا به CRC از طریق جهش ژرمینال، پایینتر از سن ابتلا به CRCایی است که بهنحوه تکگیر (Sporadic) بروز مییابد (میانگین سنی مبتلایان 35 سال)، و بهندرت در سن قبل از 20 سالگی رخ میدهد (24).
AFAP: این نوع FAP، حالتی خفیفتر از FAP کلاسیک میباشد که از طریق کاهش تعداد پولیپها (100-10)، افزایش سن ابتلا به بیماری در یک فرد، توزیع پولیپها بیشتر در سمت راست (right-sided) و کاهش سن ابتلا به CRC (بیش از7%) شناسایی میشود (25).
تعریف بالینی AFAP یکی از موضوعات بحثبرانگیز میباشد و میبایست در فرد بیمار، درحدود 99-10 آدنوما مورد بررسی قرار گیرد، هرچندکه تشخیص دقیق یک بیمار، بسیار دشوار میباشد. پیشرفت پولیپ، در بیماری AFAP که وابسته به ژن APC است، مشابه ایجاد پولیپ در بیماری MAP و یا حتی بیماری FAPای است که به شکل تکگیر ایجاد میشود. بررسی چند تن از اعضای خانواده بیمار، میتواند تعیینکننده فنوتیپ FAP باشد (26).
شکل 1: دامینهای عملکردی پروتئین MutYHو APC. A) پروتئین APC شامل یک دامین الیگومریزاسیون و یک منطقه armadillo در انتهای N، تعدادی از تکرارهای 15 و 20 آمینواسیدی در قسمت مرکزی آن و یک انتهای C است که شامل: یک دامین پایهای و محلهای باندشدن پروتئین EB1 و پروتئین دیسک انسانی بزرگ (HDLG) میباشد. B): پروتئین MutYH و دامینهای مختلف آن. اختصارات: پروتئین همانندسازی A (RPA)، ساختار مارپیچ-سنجاق سری-مارپیچ (HhH)، اندونوکلئاز آپورینیک1 (APE1)، آنتیژن هستهای تکثیر سلولی (PCNA)، ناحیه خوشهای جهش (MCR).
جدول 1: فنوتیپهای بالینی FAP وابسته به APC و پولیپوز وابسته به MutYH
FAP: پولیپوز آدنوماتوز خانوادگی، CRC: سرطان کولورکتال، AFAP: پولیپوز آدنوماتوز خانوادگی تضعیفشده، MAP: پولیپوز آدنوماتوز وابسته به MutYH، SPS: سندرم پولیپوز دندانهای، MMR : ترمیم جفت باز اشتباه
علایمی غیر از کولون: در بسیاری از بیماران مبتلا به FAP، علاوه بر کولون، بافتهای دیگر نیز درگیر میشوند و علائمی همچون پولیپهای معده و دوازدهه، تومورهای دسموئیدی (DT) و تیروئیدی و تومورهای مغزی، استخوانی (استئوما)، هایپرتروفی مادرزادی اپیتلیوم رنگدانه شبکیه، دندانهای غیرعادی و کیستهای اپیدرموئید، نیز بروز مییابند (27).
پولیپهای معده - دوازدهه: متداولترین تظاهراتی که به غیر از کولون، در این بیماران بروز پیدا میکنند، پولیپهای معده - رودهای فوقانی هستند. آنها در نواحی شکم، دوازدهه و پریآمپولار قرار میگیرند. پولیپهای معدی معمولاً از نوع پولیپهای غددی فاندیک (FGP) خوشخیم بوده و در 20% تا 84% از مبتلایان ایجاد میشوند. با این وجود، FGPهای وابسته به FAP بهطور مرسوم، بهعنوان غیرنئوپلازی در نظر گرفته میشوند و معمولاً بدون نیاز به جراحی میباشند، تاکنون مواردی از جمله دیسپلازی با درجه بالا و کارسینومای معده که ناشی از FGP بوده، در مبتلایان به FAP گزارش شده است (28,29). پولیپهای آدنوماتوی معده، در حدود 10% از پولیپهای معده حضور دارند و زمانی تشخیص داده میشوند که در محل حفرات گوارشی، به تعداد زیاد دیده شوند. علیرغم پتانسیل بدخیمی FGP، دیسپلازی معده، آدنوما و کارسینومای معده، در مبتلایان بسیار نادر میباشد (بروز کمتر از 1%). بعد از کولورکتوم، دومین و رایجترین ناحیه برای پولیپها، دوازدهه است. آدنوماهای دوازدهه در اکثر بیماران مبتلا به FAP، با ریسک تقریبی 100% ایجاد میشوند. بخشهای دوم و سوم دوازدهه، مخصوصا ناحیه پریآمپولار، دارای استعداد قابلتوجهی برای ایجاد پولیپ میباشند. این الگو احتمالا به دلیل قرارگرفتن مخاط دوازدهه در معرض اسیدهای صفراوی است که بیانگر نقش این ترکیبات در کارسینومای دوازدهه میباشد. سرطان دوازدهه با یک ریسک فزایندهی 5%، دومین علت مرگ و میر ناشی از سرطان، در بیماران مبتلا به FAP است (30,31).
نشانههای غیرکولون: هر دو نشانههای بدخیم و خوشخیم غیرکولون، در بیماران FAP شایع است. هایپرتروفی مادرزادی اپیتلیوم رنگدانه شبکیه (CHRPE) شایعترین نشانههای غیرکولون در بیماران FAP میباشد (80%-70%) که بهصورت منحنی خاکستری - قهوهای مایل به سیاه، یا ضایعات بیضیشکل در شبکیه چشم بهنظر میرسند، اما اینکه چه نوع مشکلات بالینی را ایجاد کنند، هنوز بهدرستی شناخته شده نیست. کیستهای اپیدرموئید (50%) و فیبروما (50%-25%)، بهعنوان شایعترین ضایعات زیرپوستی هستند که ممکن است موجب مشکلات ظاهری بیمار شوند. سایر نشانههای خوشخیم شامل: ناهنجاریهای دندانی (90%-79%)، پوکی استخوان (90%-50%) و تومورهای دسموئیدی (DT) (15%-10%) میباشند (34-32). تومورهای دسموئیدی، نئوپلازیهای مزانشیمی با یک رشد تدریجی هستند که دارای عدم پتانسیل متاستاتیک و در عینحال رفتار تهاجمی موضعی بوده که بهدلیل رشد سریع خود و همچنین خطر عود بالا در محل، مورد توجه محققین میباشند. ریسک ابتلا به تومور دسموئیدی (DT) در بیماران مبتلا به FAP، حدود 1000 برابر بیشتر نسبت به جمعیت عادی است. اغلب تومورهای دسموئیدی در بیماران FAP، در شکم و بیشتر در نواحی دیواره شکمی یا داخل شکمی ایجاد میشوند. از عوامل ریسک ابتلا به تومورهای دسموئیدی میتوان به سابقه جراحی شکم، یک سابقه خانوادگی مثبت برای بروز دسموئید، و همانطور که گفته شد، جایگاه جهش در ژن APC، اشاره کرد. با وجود خوشخیم بودن تومورهای دسموئیدی، آنها یکی از علل اصلی مرگ در بیماران FAP هستند (35). از بدخیمیهای غیرکولون نیز میتوان به سرطان تیروئید (3%-2%)، آدنوکارسینومای مخاطی پانکراس (1%)، هپاتوبلاستوما (1%) و تومورهای مغزی (برای نمونه، بلاستومای مغزی <1%) اشاره نمود. کارسینومای تیروئیدی پاپیلاری، سومین بدخیمی رایج در ارتباط با بیماری FAP (بعد از CRC و سرطان دوازدهه) است. با اینحال، خطر ابتلا به سرطان تیروئید، پایین بوده و بین 3%-2% با نرخ تقریبی 160 برابری نسبت به جمعیت کل تخمین زده میشود (36). زنان در ابتلا به این بیماری، دارای درصد نفوذ قابلتوجهی هستند (نسبت زن به مرد 17:1) و میانگین سنی در تشخیص آنها، 27 سال میباشد. اگرچه سرطان تیروئید در بیماران FAP میتواند گرههای لنفاوی منطقهای را نیز درگیر کند و حالت چندموضعی داشته باشد، با اینحال، پیشآگاهی مناسب، در رابطه با آن بسیار مفید است (37). بلاستومای کبدی نیز یک نئوپلازی جنینی است که عمدتا در کودکان 6 ماهه تا 3 ساله رخ میدهد، اما سن تشخیص آن میتواند از مراحل قبل از تولد تا 16 سالگی باشد. اگرچه شیمیدرمانی و جراحی، برای این مورد بسیار موفقیتآمیز است، اما تخمین زده میشود که کمتر از 25% از کل بیماران، زنده میمانند. سندرم گاردنر (Gardner) نیز بهعنوان ترکیبی از درگیریهای کولورکتال و نشانههای غیرکلون، شناخته میشود، درحالیکه سندرم تورکوت (Turcot) در ارتباط با پولیپهای کولورکتال و تومورهای مغزی معرفی میگردد (38).
همبستگی ژنوتیپ - فنوتیپ: وجود طیفی از پولیپهایی که ناشی از جهش در مناطق مختلف ژن APC اند، توسط Leppert و همکاران در سال 1990 پیشنهاد شد (39). از آن پس، مطالعات متعددی، ارتباط بین نشانههای بالینی و محل قرارگیری جهشهای ژرمینال را مشخص کردند. بهطور کلی، جهشهای بین کدونهای 178 و 309 و همینطور بین کدونهای 409 و 1580 مرتبط با فنوتیپ کلاسیک FAP، واجد بیش از 100 آدنوما هستند که مربوط به اگزونهای 8-5 و 14-9 و همچنین نیمه ابتدایی اگزون بزرگ 15 میباشند (40). بیماری FAP را میتوان براساس همبستگی ژنوتیپی - فنوتیپی، به 3 دسته تقسیمبندی کرد. 1) پولیپهای تهاجمی که دارای ویژگی شروع زودتر و تعداد بیشتر پولیپها، با جهشهایی در کدونهای 1250 تا 1464 و عمدتا در کدون 1309 هستند. 2) AFAP که معمولاً با جهشهایی در انتهای ′5 (قبل از کدون 157) و انتهای ′3 (پس از کدون 1595) ژن APC و ناحیهای از اگزون 9 (کدونهای 412-213) که بهصورت متناوب تحت پیرایش قرار میگیرند، همراه هستند و در نهایت، 3) فنوتیپ حدواسط در FAP کلاسیک، مجموع جهشهایی را شامل میشود که در باقیمانده ژن APC به خصوص انتهای ′5 بین کدون 157 و 1595 به غیر از کدون 1309، واقع شده باشند (شکل 2) (41,42). همچنین جهشهای خاصی در ژن APC، بهویژه بعد از کدون 1400، مرتبط با نشانههای غیرکولون میباشند. CHRPE مرتبط با جهشهایی است که بین کدون 311 و کدون 1456 قرار گرفتهاند و حضور تومورهای دسموئیدی نیز مرتبط با جهشهای انتهای ′3 در ژن APC و بهطور کلی پاییندست کدون 1400 (2011-1445) است. حضور پولیپهای معده و دوازدهه نیز با جهشهایی در انتهای ′3 و قبل از کدون 395 و همچنین اگزون 4 و کدون 1493-564 مرتبط است (43). سایر مواردی که نشاندهنده وجود همبستگی ژنوتیپ- فنوتیپ باشد، با شواهد اندکی مشاهده شده است. در بیماران بلاستومای کبدی، تقریباً 95% از جهشها در انتهای ′5 تا ناحیه میانی ژن APC، بین کدونهای 141 و 1751 قرار گرفتهاند. تومورهای تیروئیدی با جهشهایی مابین کدونهای 140 و 1309 مرتبط هستند (شکل 2). با وجود مشاهده همبستگی ژنوتیپ- فنوتیپ، در بین افراد بیمار و حتی بین اعضای خانواده آنها، متغیرهای قابلتوجهی وجود دارند که نشاندهنده تاثیر عوامل محیطی و یا اثر ژنهای اصلاحشده دیگر است (43,44).
شکل 2: ارتباط ژنوتیپ-فنوتیپ در ژن APC
اختصارات: DT (تومورهای دسموئیدی)، CHRPE (هایپرتروفی مادرزادی اپیتلیوم رنگدانه شبکیه)، FAP (پولیپوز آدنوماتوز خانوادگی)
الگوریتمهای تستهای ژنتیکی: قبل از آزمایشهای ژنتیک، افراد مبتلا بهمنظور فهم جوانب مثبت و منفی آزمایشات ژنتیکی سرطان، میبایست مشاوره ژنتیکی دریافت کنند. بیماران میبایست تعیین کنند که آیا از نظر روحی برای چنین تستهایی آمادگی دارند یاخیر، و سایر عوامل (مانند محرمانه بودن اطلاعات بیمار) برای آنها توضیح داده شود. هنگامی که روند مشاوره ژنتیک بهدرستی انجام شود، در صورت مشخصشدن نوع جهش ژنی، میتوان به بستگانی که دارای ریسک ابتلا به بیماری هستند، نیز نشانههای اولیه بیماری را توضیح داد. اواسط نوجوانی، زمان مناسبی برای انجام آزمایشات ژنتیک میباشد که از نظر تشخیصی و پیشگیری از سرطان دارای اهمیت بالینی قابلتوجهی است. اگر هیچ جهش پاتوژن ژرمینالی در بیمار یافت نشد، انجام آزمایشات ژنتیکی را نمیتوان به دیگر اعضای خانواده پیشنهاد کرد و فقط انجام تستهای بالینی و مراقبتهای شخصی، برای همه بستگان درجه اول فرد، توصیه میشود. روشهای متعددی برای بررسی ژن APC مورد استفاده قرار گرفته است. تعیین توالی مستقیم ژن، از تمام 15 اگزون کدکننده ژن APC بهعنوان استاندارد طلایی در شناسایی جهشهای ژنی در نظر گرفته شده است. با اینحال، روشهای دیگری نیز استفاده میشود. در گذشته چندین آزمایشگاه، از آزمایش برش پروتئین (PTT)، بر مبنای RNA استفاده میکردند. این روش براساس تجزیه و تحلیل اندازه محصولات حاصل از رونویسی و ترجمه، در شرایط آزمایشگاهی است و حساسیت آن بین 70 تا 90 درصد است. با اینحال، روش PTT دارای معایبی از جمله: دستگاههای مورد نیاز برای آزمایش و ناتوانی در شناسایی جهشهایی که در اندازه محصول تغییری ایجاد نمیکنند، میباشد. روشهای دیگر، شامل روشهای اسکنینگ (مانند: ژل الکتروفورز کنفورماسیون رشتهای) و متعاقب آن، تعیینتوالی قطعات جهشیافته میباشد. با این وجود، هیچیک از این روشها به اندازه روش تعیین توالی مستقیم ژن، حساسیت نداشته و به همین دلیل در اکثر آزمایشگاههای بالینی، بهمنظور شناسایی جهشهای نقطهای و حذفها یا اضافهشدنهای کوچک که 85 درصد از جهشهای ژن APC را تشکیل میدهند، بهعنوان روشی استاندارد مدنظر است. 15%-10% از جهشهای باقیمانده، حذفها و مضاعفشدگیهای بزرگی هستند که میتوان از طریق روشهای تکثیر لیگاند وابسته به پروب چندتایی(MLPA) ، ساترنبلات، یا PCRکمی در زمان واقعی (Real-time quantitative PCR)، آنها را شناسایی نمود (45,46). طبق توصیه دستورالعملهای کنونی، ارزیابی FAP میبایست با استفاده از تعیینتوالی کامل ژن APC انجام شود و درصورتیکه هیچگونه جهشی یافت نشد، سپس ارزیابی بازآراییهای بزرگ کروموزومی مدنظر قرار گیرد (47).
مدیریت بالینی FAP
آدنوماهای کولورکتال و CRC: هدف از مدیریت نئوپلازی کولورکتال در بیماران FAP، پیشگیری از ابتلا به CRC میباشد. این مدیریت شامل هر دو روش جراحی و پولیپکتومی آندوسکوپی (Endoscopic polypectomy) است. در خانوادههایی با FAP کلاسیک، روش سیگموئیدوسکوپی انعطافپذیر (Flexible sigmoidoscopy)، به دلیل یک توزیع تقریبا فراگیر از آدنوماها، از جمله در ناحیه رکتوم، بهعنوان تکنیک تشخیصی مناسب درنظر گرفته میشود. سن شروع غربالگری، وابسته به میزان ریسک ابتلا به آدنوماهای بدخیم کولورکتال است. با وجود اینکه بیش از 1/5% بیماران مبتلا به FAP در بین سنین 11 تا 20 سالگی به CRC دچار میشوند، اما ریسک ابتلا به آن در بیماران کمتر از 20 سال، خیلی پایین است. بنابراین غربالگری سیگموئیدوسکوپی، میبایست از سن 12 تا 14 سالگی هر دو سال یکبار انجام شود و بهصورت مادامالعمر برای افراد ناقل جهش، ادامه یابد. پس از تشخیص آدنوماها، تا زمانی که کولکتومی مشخص شده است، کلونوسکوپی کلی نیز میبایست بهطور سالانه انجام گیرد (جدول2) (35). در موارد AFAP، از زمانی که آدنوماها در سمت راست کولون واقع میشوند، بهجای سیگموئیدوسکوپی، کلونوسکوپی توصیه میشود. در اینحالت، بهمنظور شروع تشخیص پولیپوز، از سن 18 تا 20 سالگی میبایست هر دو سال یکبار غربالگری صورت گیرد و هنگامی که آدنوماها شناسایی شوند، کلونوسکوپی میبایست بهصورت سالانه انجام پذیرد (48). بهمنظور جلوگیری از بروز و مرگ و میر ناشی از CRC پیشرفته، عمل جراحی حذفی کلون، در مرحله پیش از بدخیمی، حائز اهمیت است. در نوع کلاسیک FAP، زمانی که پولیپوز شدت مییابد، یا پولیپهای آزاردهنده (ایجاد زخم cm1 با درجه بالای دیسپلازی) شناسایی شوند، معمولاً کولکتومی بهمنظور پیشگیری از CRC توصیه میشود. اکثر بیماران مبتلا به FAP کلاسیک، بین سنین 15 تا 25 سالگی تحت عمل جراحی قرار میگیرند. راه درمانی AFAP نیز معمولاً آندوسکوپی است و در صورتی که این امر میسر نباشد، عمل جراحی بهشیوهای مشابه با FAP کلاسیک انجام میگیرد. گزینههای جراحی شامل: پروکتولکتومی با آناستوموز ایلئوآنال (IPAA) و کولکتومی توتال با آناستوموز ایلئورکتال (IRA) است. IPAA در مقایسه با IRA با کمترین میزان عوارض و معمولاً عملکرد خوب روده پس از جراحی، نسبتاً سادهتر میباشد. در مورد IPAA، جراحی گستردهتری (از جمله مداخله در لگن) موردنیاز است که منجر به کاهش باروری و بدترشدن عملکرد رودهها نیز میگردد (49). انتخاب روش جراحی، عمدتا وابسته به سن تشخیص، دسموئیدها، باروری و تعداد پولیپهای رکتال (15 تا 20 پولیپ) و همچنین تصمیم بیمار پس از دریافت اطلاعات جامع در مورد مزایا و خطرات هر کدام از روشهای درمانی میباشد (50). برخی از محققین، استفاده از شواهد مربوط به ارتباط ژنوتیپ- فنوتیپ را بهعنوان راهنما، در جراحی بیمارانی با رکتومی نسبتا نازک پیشنهاد کردهاند (43). IPAA ممکن است در بیمارانی با ژنوتیپ تشدیدیافته نیز توصیه شود، زیرا این بیماران در معرض خطر ابتلا به پولیپوز شدید راست - روده هستند که درصورت انجام IRA، به پروکتکتومی مجدد نیاز خواهند داشت. پس از جراحی، برای آندسته از بیماران دارای بقایای مقعدی، پیگیری از طریق آندوسکوپی بهدلیل وجود ریسک ابتلا به سرطان رکتال (بیش از 30% موارد) توصیه میشود. در بسیاری از مطالعات نشان داده شده است که پس از پروکتوکلکتومی (Proctocolectomy) مجدد نیز آدنوماها و گاهی اوقات حتی آدنوکارسینوماها در کیسه ایلئوس مقعد دیده شدهاند. بنابراین مراقبت بر کیسه و منطقه مقعدی که دستخوش تغییر شده است، ضرورت دارد (51). بهطور کلی، در رابطه با نشانههای غیرکلون، غربالگری میبایست در زمان شروع تشخیص پولیپها یا بین سنین 25 تا 30 سالگی صورت پذیرد. در صورت شناسایی آدنوماها، آندوسکوپی معده – دوازدهه در هر دو جهت جلویی و جانبی (به منظور رویت صحیح آمپولاواتر: Vater’s ampulla) میبایست هر پنج سال یکبار انجام گیرد. در عمل، با توجه به شیوع کم آدنوکارسینومای معدی، مراقبت از طریق آندوسکوپی دستگاه گوارش فوقاتی، بهدلیل وجود ریسک ابتلا به سرطان دوازدهه ضروری است. معده بهعنوان بخشی از این مراقبت، رویت میشود، اما بیوپسی یا پولیپکتومی (Polypectomy) تنها برای ضایعات بزرگ و غیرمعمول، بهخصوص در آنتروم، اعمال میگردد (52). به منظور استانداردسازی و مدیریت پولیپهای دوازدهه در بیماران FAP، Spigelman و همکارانش، یک سیستم طبقهبندی را براساس چهار متغیر پیشآگاهی: تعداد پولیپها، اندازه آنها، بافتشناسی و درجه دیسپلازی معرفی کردند (جدول 3) (53). در مرحله I (با 4 امتیاز) حدمتوسطی از بیماری مشاهده میشود و در مراحل III و IV (با امتیاز بالاتر از 6) پولیپهای وخیمی در دوازدهه، با ریسک قابلتوجهی برای ابتلا به سرطان دوازدهه (7 تا 36 درصد)، بروز مییابند. تقریبا 80% بیماران، در مرحله I تا III و 10 تا 20 درصد موارد، در مرحله IV بیماری هستند. شواهد موجود حاکی از آن است که معاینه دوازدهه، از طریق کرومواندوسکوپی (Chromoendoscopy) یا تصویربرداری موجب افزایش تشخیص آدنوماهای دوازدهه میگردد، اما منجر به تغییر قابلملاحظهای در مراحل Spigelman نمیشود. مدیریت بیمارانی با چندین آدنومای بزرگتر (مرحله III یا بالاتر) ، چالشبرانگیز بوده و میبایست در مراکز بالینی اختصاصی انجام شود (54). میزان عود پیشرفت آدنوما، بعد از درمان آندوسکوپی نیز بسیار زیاد است (بیش از 50%) و درمان این موارد نیز با عوارض خطرناکی همچون خطر سوراخشدن، خونریزی و آسیب پانکراتیک همراه است (52(. از آنجاییکه حذف تمام آدنوماها امکانپذیر نیست، اولویت اول، حذف آدنوماهای بزرگ (بیش از cm1) یا آدنوماهایی با درجه دیسپلازی بالا، با هدف به تاخیرانداختن یا اجتناب از عمل جراحی، میباشد. در بیمارانی با درجه IV وخامت، جراحی غالبا ضروری است که شامل: دئودنوتومی (Duodenotomy) با پولیپکتومی، دئودنکتومی پانکراس و پانکراتکتومی دوازدهه میباشد (55).
جدول 2: توصیههای مدیریت بالینی برای FAP
اختصارات: پولیپوز آدنوماتوز خانوادگی:FAP، توموگرافی رایانهای : CT، تصویربرداری رزونانس مغناطیسی : MRI. پولیپهای معده- دوازدهه
جدول3: رده بندی بر اساس معیارهای Spigelman
مرحله بندی بر اساس امتیاز: مرحله 0: 0 امتیاز، مرحله I: 4 امتیاز، مرحله II: 6-5 امتیاز، مرحله III: 8-7 امتیاز، مرحله IV: 12-9 امتیاز.
مدیریت بالینی سایر تومورها: با توجه به افزایش خطر ابتلا به سرطان تیروئید، متخصصین بر این باورند که بررسی گردن و یا تیروئید، باید از سن 30-25 سالگی و بهصورت سالانه انجام شود. پیشرفت تومور دیسموئید (DT) نیز عمدتا وابسته به یک سابقه خانوادگی مثبت، جراحی شکم، و محل بروز جهش بوده و میتواند در داخل یا دیواره شکمی ایجاد شود. DT را میتوان از طریق توموگرافی رایانهای (CT) یا تصویربرداری رزونانس مغناطیسی (MRI) تشخیص داد. گزینههای درمانی آن عبارتند از: درمان دارویی (داروهای ضدالتهاب غیراستروئیدی NSAID و یا آنتیاستروژنها)، شیمیدرمانی، مداخله جراحی و یا پرتودرمانی (56). طبق شواهد بالینی، اثرگذاری این درمانها نسبتاً ضعیف بوده و مبتنی بر مطالعات محدودی است. بااینحال، در صورت عدم بروز عوارض و به دلیل بالابودن میزان عود DT، هرگونه مداخله از طریق عمل جراحی تومورهای داخل شکمی، میبایست به تعویق بیفتد. متخصصین در بیمارانی با DTهای بزرگ یا در حال رشد، داروهای تاموکسیفن با سولینداک (Sulindac) را بهعنوان خط اول درمان توصیه میکنند و هنگامیکه بیمارانی با تومورهای داخل شکمی به این درمان پاسخ نمیدهند، شیمیدرمانی یا پرتودرمانی را تجویز میکنند. تومورهای دسموئیدی واقع در دیواره شکمی و DTهای بافت مزانتریک، میبایست بهطرز متفاوتی از هم، مدنظر قرار گیرند. عمل جراحی معمولاً بهعنوان خط اول درمان، برای مداوای DTهای دیواره شکمی مورد استفاده قرار میگیرد. با اینحال، در مورد DT مزانتریک با توجه به شدت بیماری، عوارض احتمالی و سنجیدن مزیتهای روش درمان، نسبت به ریسک روشهای دیگر، استراتژی بهینه بهصورت شخصی انتخاب میشود (57). سایر بدخیمیهای غیر رودهای (پانکراتیک، مغزی و آدرنال) دارای شیوع بسیار کمتری میباشند که استفاده از مطالعات پرهزینه دیگری برای آنها توصیه نمیشود. با اینحال، تستهای نظارتی در بیمارانی، با یک سابقه خانوادگی قوی، در رابطه با هریک از این علایم خاص غیر رودهای و آن دسته از افرادیکه دارای نشانههای منتسب به این بیماریاند، میبایست انجام شود.
روشهای پیشگیری شیمیایی: در بیماران FAP بهمنظور پیشگیری، از روشهای شیمیایی، نظیر استفاده از داروهای NSAID نیز استفاده میشود. NSDAIها با عملکرد آنزیم COX-1 و COX-2 (Cyclooxygenase) در تداخل هستند و از تبدیل اسید آراشیدونیک به پروستاگلاندین¬ها که در پیدایش درد نقش دارند، جلوگیری می¬کنند (58). سولینداک نیز بهعنوان یکی از اولین داروهایی است که در این بیماری موثر بوده است. استفاده طولانیمدت از این دارو، منجر به کاهش 50 درصدی آدنوماهای کولورکتال در کولون و همچنین در رکتوم بیماران پس از کولکتومی میشود، اما بر پولیپوز دوازدهه موثر نیست. با اینوجود، داروی سالینداک از پیشرفت آدنوماها در بیماری FAP جلوگیری نمیکند (59). داروی سلکوکسیب (Celecoxib) نیز بهعنوان مهارکننده انتخابی پروتئین (cyclooxygenase-2) COX-2، همراه با عوارض گوارشی کمتر نسبت به داروی سالینداک، و بهمنظور کاهش 28 درصدی تعداد آدنوماهای کولورکتال معرفی شده است و توانسته است تعداد آدنوماهای دوازدهه را در بیماران، کاهش دهد. با اینوجود، در مصرفکنندگانی که بهصورت طولانیمدت از مهارکننده انتخابی COX-2 دیگری (مانند روفکوکسیب: Rofecoxib) استفاده میکردند، عوارض قلبی- عروقی (انفارکتوس یا سکته قلبی) گزارش شده است و بنابراین نقش و اثر این داروها، همچنان مورد بحث باقی میماند و میبایست تنها در بیماران انتخابشدهای که بدون فاکتورهای خطر و ریسک قلبی - عروقی هستند، مدنظر قرار گیرند (60). با این وجود، NSAIDها (سولینداک و سلکوکسیب) را نمیتوان جایگزین عمل جراحی در بیماران مبتلا به FAP کولون نمود و احتمالاً نقش آنها در به تعویق انداختن عمل جراحی، در مبتلایان به پولیپوز کولون یا بیمارانی با پولیپوز رکتال پس از کولکتومی میباشد. در رابطه با پولیپوز دوازدهه، بهاین دلیل که گزینههای درمانی، اعم از اندوسکوپی و جراحی در برخی موارد همراه با عوارض چشمگیری هستند، مصرف داروی سلکوکسیب برای بیماران مبتلا به پولیپوز شدید دوازدهه (مرحله III یا IV)، امری قابل قبول میباشد. اگرچه داروی سلکوکسیب برای درمان بیماری FAP در چندین کشور مورد تایید واقع شده است، اما برخی متخصصین به دلیل اثرات و عوارض قلبی - عروقی، نسبت به تجویز آن بیرغبت بوده و در نتیجه این دارو بهندرت مورد استفاده قرار میگیرد (61).
:MAP MAP یک اختلال ارثی با الگوی اتوزومال مغلوب است که به علت جهشهای ژرمینال دوآللی در ژن MutYH ایجاد میشود. این بیماری برای اولینبار در سال 2002 توسط AL-Tassan و همکارانش، در یک خانواده بریتانیایی با سه عضو مبتلا و توارث مغلوب آدنوماهای متعدد کولورکتال و کارسینوما، مورد بررسی قرار گرفت. علایم بالینی در بیماران مبتلا به MAP، دارای تنوع زیادی میباشد، اما معمولاً بهصورت یک فنوتیپ پولیپوز تضعیفشده، همراه با تعداد کمتر از 100 آدنوما بروز مییابد. برخی از بیماران، به علایمی غیر از روده نیز دچار میشوند که در بیماران FAP غیرقابل تشخیص است (62).
ژن MutYH: ژن MutYH بر روی کروموزوم 1p34.1 واقع شده و دارای 16 اگزون کدکننده یک پروتئین 535 آمینواسیدی میباشد (شکل 1B). این ژن یک عضو از سیستم ترمیمی برش بازی (BER) را کد میکند. این سیستم، متشکل از 3 آنزیم (MutYH, OGG1, MTH1) میباشد که در محافظت سلولها در برابر اثرات جهشزای متابولیسم هوازی و بهویژه اکسیداسیون نوکلئوتید گوانین که منجر به تشکیل 8-اکسوگوانین (oxoG-8: (8-hydroxyguanine, 8-oxo-Gua, or OH8Gua میگردد، شرکت میکند. پروتئین MutYH همرا با OGG1 و MTH1 از بروز جهشهای سوماتیک که ناشی از 8-اکسوگوانین بوده و تمایل زیادی به جایگزینی نوکلئوتید آدنین (A) به جای سیتوزین دارند، ممانعت بهعمل میآورد. بهویژه آنکه پروتئین MutYH مسئول حذف آدنینهایی است که به اشتباه با 8-اکسوگوانین جفت شدهاند (63). در غیاب یک کپی عملکردی از پروتئین MutYH به دلیل جهشهای دو آللی، زمانی که یک 8-اکسوگوانین، بهصورت جفت باز اشتباه در الگوی DNA قرار گیرد، در دور بعدی همانندسازی جفت بازG:C به T:A تبدیل میشود. به همین علت، جهشهای سوماتیکG:C به T:A در ژنهایی مانند: APC یا KRAS اغلب در آدنوماها و تومورهای مرتبط با MutYH رخ میدهد. بهعنوان مثال، در ژن KRAS (جهش c.34G>T در کدون 12) اغلب در بیماران MAP مبتلا به CRC بیشتر است (64%). بنابراین آنالیز جهشهای سوماتیک در ژن KRAS بهعنوان یک تست پیشغربالگری برای شناسایی بیماران CRC که واجد شرایط برای آزمایشات ژنتیکی در مورد جهشهای ژرمینال ژن MutYH هستند، توصیه شده است (64). از آنجاکه در بیماران مبتلا به MAP آدنوماهای معمولی و همچنین پولیپهای دندانهای (پولیپهای هایپرپلازی) میتوانند حضور داشته باشند، وجود دو مسیر مجزا پیشنهاد شده است که یکی از آنها، بروز جهشهایی در ژن KRAS و یا ژن APC است که منجر به آدنوماهای معمولی شده و مسیر بعدی، بدون رخداد جهش در ژن APC و فقط از طریق جهشهایی در ژن KRAS است که منجر به پولیپهای هایپرپلازی میشود (65).
نگاهی به جهشهای ژن MutYH: تاکنون بیش از 300 واریانت منحصر بهفرد و قریب به 80 جهش بیماریزا در این ژن شناسایی شده است. اکثر آنها از نوع جایگزینیهای بدمعنی و تعداد کمی از جهشها نیز از نوع کوتاهکننده یا جایگاه پیرایش هستند. اگرچه در موارد نادری، جهشهایی از نوع حذفهای ژنومی بزرگ، تغییر چارچوب و جهشهای بیمعنی نیز گزارش شده است. جهشها تقریبا در تمامی اگزونها (به غیر از اگزون 1 و 2) گزارش شده است. غالبترین جهشهای بدمعنی، دو جهش واقع در نقاط داغ ژن میباشند که شامل: p.Y179C (c.536A>G; p.Tyr179Cys) در اگزون 7 و جهش p.G396D (c.1187G>A; p.Gly396Asp) در اگزون 13 هستند و حدود 70 تا 80 درصد از تمام جهشهای جمعیتهای اروپایی را در بر میگیرند (جدول 4) (66). مطالعات گذشته، اختلافات جغرافیایی و نژادی را در مورد فراوانی جهشهای ژن MutYH را بررسی نمودهاند. همانطور که ذکر شد، دو جهش p.Y179C و p.G396D در جمعیتهای اروپایی، بهمراتب شایعترین واریانتهای بیماریزا میباشند. علاوه براین، با توجه به تفاوتهای قومی و جغرافیایی، واریانتهای مختلف، دارای نقش اثرگذاری در سایر جمعیتها هستند، بهعنوان مثال: جهش بیمعنی p.Y104X در بیماران پاکستانی و جهش بیمعنی p.E480X در بیماران هندی بسیار حائز اهمیتاند (67). جهشهای خاص دیگری نیز در جمعیتهای ژاپنی و جنوب اروپا گزارش شده است (جدول 4). مطالعات متعددی به بررسی فراوانی دو نوع متداول از جهشهای بدمعنی (p.Y179C و p.G396D) پرداختهاند و پیش بینی شده است که تقریبا 1 تا 2 درصد از جمعیت عمومی (با منشا اروپایی) ناقل این جهشها باشند (68).
جدول 4: جهشهای شایع ژرمینال در ژن MutYH در جمعیتهای مختلف
فنوتیپ: طیف بالینی جهشهای ژرمینال در ژن MutYH هتروژنوس بوده و میتواند گستره وسیعی از فنوتیپها را شامل شود.
پولیپوز آدنوماتوی کلاسیک و تضعیفشده: اکثر ناقلین جهش دو آللی در ژن MutYH دارای ده تا چند صد پولیپ هستند و در تعداد بسیار کمی از بیماران بیش از 500 پولیپ نیز گسترش یافتهاست. در ابتدا، جهشهای دو آللی در یک سوم از بیماران AFAP با APC منفی، همراه با بیش از 15 آدنوما و در حدود 10 درصد بیماران مبتلا به FAP کلاسیک با APC منفی و بهویژه، در آن دسته از مواردی که دارای یک الگوی توارثی مغلوب بودند، شناسایی شد (69). طی مطالعه دیگری نیز در مورد جهشهای دو آللی در ژن MutYH، مشخص گردید که این جهشها در 95 از 119 بیمار با بیش از 1000 آدنوما و 94 از 1338 بیمار با 100 تا 999 آدنوما و 233 از 3253 بیمار با 20 تا 99 آدنوما و 37 از 970 بیمار با 10 تا 19 آدنوما مشاهده شد (70). سندرم پولیپوز پایهدار و بیماران مبتلا به پولیپهای چندگانه آدنوما و پولیپهای پایهدار، بهطور شایع در MAP یافت میشوند. در تعداد کمی از بیماران MAP، بهمنظور بررسی پولیپوز آدنوماتوز، از معیارهای WHO برای سندرم پولیپوز پایهدار استفاده میشود (زمانی که یک بیمار حداقل دارای یکی از معیارهای زیر باشد: 1- وجود حداقل پنج پولیپ پایهدار در نزدیکی کولون سیگموئید که دو تا از آنها دارای قطری بیش از mm 10 باشند، 2- هریک از پولیپهای پایهداری که نزدیک کولون سیگموئید فردی با پولیپوز پایهدار درجه اول ایجاد شود 3- بیش از 20 پولیپ پایهدار که در اندازههای متفاوت در سرتاسر کولون توزیع شده باشند) (71). در مطالعهای که بر روی بیماران شناسایی شده با حداقل ده پولیپ از هر نوع بافتی (شامل پولیپهای آدنوماتوز و پایهدار) متمرکز شده بود، 6/7% (405/27) از بیماران، بهصورت ناقلین جهش دو آللی در ژن MutYH مشاهده شدند (71).
CRC بدون پولیپوز: در مطالعات CRC مبتنی بر جمعیت که در آن، بیماران بر اساس تشخیص CRC مورد بررسی قرار گرفته بودند، جهشهای دو آللی در ژن MutYH در 2 تا 3 درصد جمعیت یافت شد. در چندین مطالعه دیگر مبتنی بر جمعیت، بیش از یک سوم ناقلین جهش دو آللی در ژن MutYH، دارای فنوتیپ پولیپوز نبودند. بر ایناساس، جهشهای دو آللی در ژن MutYH همیشه با یک فنوتیپ پولیپوز همراه نیستند، بنابراین، این گونه جهشها میبایست در بیماران مبتلا به CRC زودهنگام (قبل از سن 50 سالگی) در نظر گرفته شوند، بهویژه اگر احتمال مبتلا بودن به سندرم لینچ در بیماران، رد شده باشد. بااینحال، فنوتیپهای غیر معمول نیز گزارش شده است (72). اخیرا در یک مطالعه، ژن MutYH در 85 مورد مشکوک به سندرم لینچ، همراه با تومورهایی مشاهده شدند که نشاندهنده نقص سیستم ترمیمی جفت باز اشتباه و همچنین بدون هیچ جهش ژرمینال APC قابل تشخیص بودند، این بیماران ناقل جهش دوآللی (p.Y179C) همراه با CRC گسترشیافته، کارسینومای اوروتلیال و کارسینومای غدد چربی، شناسایی شدند. در این بیماران خاص، تعیینتوالی تومور، دو جهش سوماتیکی در سیستم ترمیم جفت باز را نشان داد که به اینترتیب فنوتیپ بیماری قابل توضیح بود (73).
جهشهای هتروژنوس در ژن MutYH و خطر ابتلا به CRC: ریسک ابتلا به CRC در افراد دارای جهشهای یکآللی (هتروزیگوت) بهعنوان مبحثی مهم و چالشبرانگیز بوده است. چندین متاآنالیز، همراه با OR (نسبت احتمالات) یا RR (نسبت نسبی) بین بیماران و افراد عادی جمعیتهای مختلف انجام شد که بیشتر آنها هیچ گونه تفاوت آماری را نشان ندادند. اخیراً طبق جدیدترین یافتهها، مشخص شده است که فراوانی ناقلین جهشهای یکآللی که دارای یک سابقه خانوادگی از بیماری بودند، نسبت به افراد کنترل (P=0.02) بیشتر بوده و پیشنهاد میشود که ناقلین جهشهای یکآللی با سابقه خانوادگی CRC، نیز ممکن است در معرض خطر بیشتری باشند (74,75).
علایم و نشانههای غیرکولون: از آنجاییکه استرس اکسیداتیو در بافتهای مختلف وجود دارد، میتوان انتظار داشت که یک ژن MutYH معیوب، باعث ایجاد نئوپلازیهایی در سایر اعضای بدن نیز گردد. Vogt و همکارانش اخیرا طیفی از خصوصیات غیرکولون گروه بزرگی از بیماران MAP را (276 بیمار مبتلا به MAP) تعیین نمودند. در این مطالعه 17% بیماران، در معرض خطر ابتلا به پولیپوز دوازدهه در طول مدت عمر خود بوده و 4% ریسک ابتلا به سرطان دوازدهه و 38% در معرض خطر ابتلا به هر یک از سرطانهای غیر از روده میباشند. بروز بدخیمیهای غیر رودهای، در بین بیماران تقریبا دو برابر بیشتر از جمعیت کلی بوده و افزایش قابلتوجهی در بروز سرطانهای تخمدان، مثانه و پوست، و روندی افزایشی در ریسک ابتلا به سرطان پستان برای آنها وجود دارد (76). سن متوسط در تشخیص سرطانهایی غیر از روده، بین سنین 51 تا 61 سال متغیر است. سایر عوارض دیده شده در بیماران FAP اعم از FGP معده، لیپوما، CHRPE، کیست اپیدرموئید، DTو کارسینومای تیروئیدی، در شمار اندکی از بیماران MAP نیز گزارش شده است. بهطورکلی، میزان بروز علایم مرتبط با FAP، در بیماران MAP با نسبت کمتری دیده میشود (77).
همبستگی ژنوتیپ- فنوتیپ: همبستگی ژنوتیپ- فنوتیپ، همانند ژن APC در جهشهای ژن MutYH نیز توصیف شده است. برخی مطالعات نشان دادهاند که طبق بررسیهای ژنتیکی، جهش p.Y179C نسبت به جهش نقطهای G396D، منجر به کاهش هرچه بیشتر فعالیت گلیکولازی پروتئین MutYH میگردد و در بیمارانی با جهشهای دو آللی Y179C نسبت به جهشهای G396D، با یک فنوتیپ شدیدتری (بهعنوان مثال،شروع زودتر و تعداد بیشتر پولیپها) همراه است (71). در مطالعه دیگری نیز، واریانت p.G396D همراه با پیشرفت پولیپهای پایهدار در بیماران MAP گزارش شده است (78).
الگوریتمهای تستهای ژنتیکی: بیمارانی که دارای یک شکل تضعیف شدهای از پولیپوز آدنوماتوز یا FAP کلاسیک با یک الگوی توارثی مغلوب هستند، مشکوک به جهشهای دو آللی در ژن MutYH میباشند. همچنین در بیماران مبتلا به CRC تا قبل از سن 50 سالگی و در بیماران دارای پولیپهای متعدد کولون (بیشتر از 10 عدد، از جمله آدنوماتوز و پایهدار) باید بیشتر مدنظر قرار گیرند. هنگام یافتن یک فرد ناقل جهشهای دو آللی در ژن MutYH، آزمایشات ژنتیکی را میتوان برای بستگان درجه اول او نیز پیشنهاد داد، بهویژه خواهر و برادرهایی که به میزان 25 درصد دارای ریسک ناقلبودن جهشهای دو آللی هستند. در بیماران MAP احتمال ریسک ابتلا به این بیماری در فرزندان آنها، بستگی به وضعیت ژنتیکی همسر آن فرد بیمار دارد. آنالیز جهشهای ژرمینال، معمولا شامل دو نوع از متداولترین جهشها (p.G396D و p.Y179C) میباشد. تعیینتوالی کامل ژن در موارد زیر توصیه میشود: 1) افرادی که ناقل یک یا دو جهش متداول هستند، 2) افرادی که از نظر نژادی هندی و اروپایی نیستند و 3) در افرادی با نژاد هندی و اروپایی با سابقهی خانوادگی که برای جهشهای شایع، منفی بوده باشند. از آنجا که حذفها و مضاعفشدگیهای بزرگ، بهطور فوقالعادهای در ژن MutYH گزارش شدهاند، دیگر نیازی به انجام روشهایی برای آنالیز این جهشها نیست (63).
مدیریت بالینی: پروتکل مراقبتی پیشنهادشده برای هر دو حالت بیماران MAP و AFAP مشابه میباشد. افراد میبایست از سن 18 تا 20 سالگی کولونوسکوپی را شروع کرده و هر دو سال یکبار و بهصورت مادامالعمر آن را ادامه دهند. مدیریت پولیپهای کولورکتال، شبیه آن چیزی است که برای بیماران AFAP گزارش شده است. در برخی از بیماران، به دلیل فنوتیپ تضعیفشده، احتمال از بینرفتن این پولیپها در طی آندوسکوپی وجود دارد. با اینحال، در صورت نیاز به جراحی، تصمیماتی مشابه با AFAP اتخاذ میشود. در بیماران مبتلا به MAP، آندوسکوپی نواحی فوقانی، همانند آنچه که برای بیماران AFAP بیان شده است، در سنین 25 تا 30 سالگی توصیه میشود. در این شرایط، هیچگونه شواهدی مبنی بر مفیدبودن روشهای پیشگیری با داروهای شیمیایی وجود ندارد (62).
نتیجهگیری
FAP یک سندرم اتوزومال غالب است که مسئول تقریباً 1٪ موارد CRC محسوب میشود. یک بیمار مبتلا به FAP احتمال بسیار بالایی برای ابتلا به CRC دارد، اما ممکن است سایر تظاهرات و بدخیمیهای خارج کولونی را نیز بروز دهد. این امر، اهمیت تشخیص زودهنگام و دقیق و نظارت فعال بیماری را نشان میدهد، بنابراین بهبود بیشتر ابزارهای تشخیصی را ضروری میکند. مطالعات متعددی بهطور خاص برای تهیه پانلهای چندژنی در تشخیص FAP انجام شده است و تلاش برای شناسایی واریانتهای ژنی که در حالحاضر ناشناخته ماندهاند، ادامه دارد. FAP غالباً در اثر جهشهای نوکلئوتیدی در ژن APC (کروموزوم 5q21) ایجاد میشود. یک شکل تضعیفشده از FAP نیز وجود دارد که با ایجاد کمتر از 100 آدنومای کولورکتال و شروع تاخیری CRC مشخص میشود. پولیپوز وابسته به MutYH (MAP)، الگوی اتوزومال مغلوب دارد و ناشی از جهشهای نوکلئوتیدی در ژن MutYH است. نظارت دقیق آندوسکوپی برای همه بیماران FAP، MAP و اعضای خانواده در معرض خطر آنها، توصیه میشود. علاوه بر این، تحقیقات بیشتر در تعیین اهمیت تظاهرات و بدخیمیهای خارج کولونی FAP و ارتباط آن با سایر تظاهرات بیماری، ممکن است اطلاعاتی را فراهم کند که بر استراتژیهای مراقبتی و درمانی، در آینده تأثیرگذار باشد. در نهایت، هدف همه این تحقیقات، کاهش عوارض بیماری و تلاش جهت افزایش طول عمر و کیفیت زندگی قابل قبول برای افراد آسیبدیده است.
سپاسگزاری
نویسندگان مقاله از حمایتهای دانشگاه یزد در انجام این پژوهش تشکر و قدرانی میکنند.
حامی مالی: ندارد.
تعارض در منافع: وجود ندارد.
مشارکت نویسندگان
محمد مهدی حیدری در ارائه ایده، مهری خاتمی در طراحی مطالعه، الهام افخمی عقدا و مریم طهماسبی در جمعآوری دادهها، زهرا شاکر اردکانی در تجزیه و تحلیل دادهها مشارکت داشته و همه نویسندگان در تدوین، ویرایش اولیه و نهایی مقاله و پاسخگویی به سوالات مرتبط با مقاله سهیم هستند.
References:
1- Ditonno I, Novielli D, Celiberto F, Rizzi S, Rendina M, Ierardi E, et al. Molecular Pathways of Carcinogenesis in Familial Adenomatous Polyposis. Int J Mol Sci 2023; 24(6): 5687.
2- Half E, Bercovich D, Rozen P. Familial Adenomatous Polyposis. Orphanet J Rare Dis 2009; 4: 22.
3- Claes K, Dahan K, Tejpar S, De Paepe A, Bonduelle M, Abramowicz M, et al. The Genetics of Familial Adenomatous Polyposis (Fap) and Mutyh-Associated Polyposis (Map). Acta Gastroenterol Belg 2011; 74(3): 421-6.
4- Heidari MM, Ebrahimi F, Shaker Ardakani Z, Mirzaei M, Mirhosseini S, Khatami M. Molecular Study of Nucleotide Changes of Atpase6 and Mt-Cyb Genes in the Mitochondrial Genome of Patients with Familial Adenomatous Polyposis (Fap). The Journal of Shahid Sadoughi University of Medical Sciences 2023; 31 (5): 6632-45.
5- Afkhami E, Heidari MM, Khatami M, Ghadamyari F, Dianatpour S. Detection of Novel Mitochondrial Mutations in Cytochrome C Oxidase Subunit 1 (Cox1) in Patients with Familial Adenomatous Polyposis (Fap). Clin Transl Oncol 2020; 22(6): 908-18.
6- Heidari MM, Afkhami E, Khatami M, Farzaneh G. Genetic Analysis of D-Loop Region of Mitochondrial Dna Sequence in Iranian Patients with Familial Adenomatous Polyposis (Fap): A Case-Control Study. Journal of Rafsanjan University of Medical Sciences 2023; 21(12): 1307-22.
7- Shaker Ardakani Z, Heidari MM, Khatami M, Bitaraf Sani M. Association of Pathogenic Missense and Nonsense Mutations in Mitochondrial Coii Gene with Familial Adenomatous Polyposis (Fap). Int J Mol Cell Med 2020; 9(4): 255-65.
8- Parker TW, Neufeld KL. Apc Controls Wnt-Induced Β-Catenin Destruction Complex Recruitment in Human Colonocytes. Sci Rep 2020; 10(1): 2957.
9- Hankey W, Frankel WL, Groden J. Functions of the Apc Tumor Suppressor Protein Dependent and Independent of Canonical Wnt Signaling: Implications for Therapeutic Targeting. Cancer Metastasis Rev 2018; 37(1): 159-72.
10- Pino MS, Chung DC. The Chromosomal Instability Pathway in Colon Cancer. Gastroenterology 2010; 138(6): 2059-72.
11- Kapitanović S, Cacev T, Radosević S, Spaventi S, Spaventi R, Pavelić K. Apc Gene Loss of Heterozygosity, Mutations, E1317q, and I1307k Germ-Line Variants in Sporadic Colon Cancer in Croatia. Exp Mol Pathol 2004; 77(3): 193-200.
12- Nielsen M, Hes FJ, Nagengast FM, Weiss MM, Mathus-Vliegen EM, Morreau H, et al. Germline Mutations in Apc and Mutyh are Responsible for the Majority of Families with Attenuated Familial Adenomatous Polyposis. Clin Genet 2007; 71(5): 427-33.
13- Balmaña J, Balaguer F, Cervantes A, Arnold D; ESMO Guidelines Working Group. Familial Risk-Colorectal Cancer: Esmo Clinical Practice Guidelines. Ann Oncol 2013; 24 Suppl 6: vi73-80.
14- Ghadamyari F, Heidari MM, Zeinali S, Khatami M, Merat S, Bagherian H, Rejali L. Mutational Screening through Comprehensive Bioinformatics Analysis to Detect Novel Germline Mutations in the Apc Gene in Patients with Familial Adenomatous Polyposis (Fap). J Clin Lab Anal 2021; 35(5): e23768.
15- Rowan AJ, Lamlum H, Ilyas M, Wheeler J, Straub J, Papadopoulou A, et al. APC Mutations in Sporadic Colorectal Tumors: A Mutational "Hotspot" and Interdependence of the "Two Hits" . Proc Natl Acad Sci U S A 2000; 97(7): 3352-7.
16- De Queiroz Rossanese LB, De Lima Marson FA, Ribeiro JD, Coy CS, Bertuzzo CS. Apc Germline Mutations in Families with Familial Adenomatous Polyposis. Oncol Rep 2013; 30(5): 2081-8.
17- Heinen CD. Genotype to Phenotype: Analyzing the Effects of Inherited Mutations in Colorectal Cancer Families. Mutat Res 2010; 693(1-2): 32-45.
18- Schneikert J, Behrens J. The Canonical Wnt Signalling Pathway and its Apc Partner in Colon Cancer Development. Gut 2007; 56(3): 417-25.
19- Dallosso AR, Jones S, Azzopardi D, Moskvina V, Al-Tassan N, Williams GT, et al. The Apc Variant P.Glu1317gln Predisposes to Colorectal Adenomas by a Novel Mechanism of Relaxing the Target for Tumorigenic Somatic Apc Mutations. Hum Mutat 2009; 30(10): 1412-8.
20- Disciglio V, Forte G, Fasano C, Sanese P, Lepore Signorile M, De Marco K, et al. Apc Splicing Mutations Leading to in-Frame Exon 12 or Exon 13 Skipping are Rare Events in Fap Pathogenesis and Define the Clinical Outcome. Genes(Basel) 2021; 12(3): 353.
21- Schwab AL, Tuohy TM, Condie M, Neklason DW, Burt RW. Gonadal Mosaicism and Familial Adenomatous Polyposis. Fam Cancer 2008; 7(2): 173-77.
22- Aitchison A, Hakkaart C, Day RC, Morrin HR, Frizelle FA, Keenan JI. Apc Mutations are Not Confined to Hotspot Regions in Early-Onset Colorectal Cancer. Cancers(Basel) 2020; 12(12): 3829.
23- Fearnhead NS, Britton MP, Bodmer WF. The Abc of Apc. Hum Mol Genet 2001; 10(7): 721-33.
24- Talseth-Palmer BA. The Genetic Basis of Colonic Adenomatous Polyposis Syndromes. Hered Cancer Clin Pract 2017; 15: 5.
25- Sant V, Reich E, Khanna L, Cao W, Kornacki S, Grucela A.. Attenuated Familial Adenomatous Polyposis (Afap) in a Patient Associated with a Novel Mutation in Apc. BMJ Case Rep 2019; 12(11): e231232.
26- Ibrahim A, Barnes DR, Dunlop J, Barrowdale D, Antoniou AC, Berg JN. Attenuated Familial Adenomatous Polyposis Manifests as Autosomal Dominant Late-Onset Colorectal Cancer. Eur J Hum Genet 2014; 22(11): 1330-3.
27- Latchford A, Volikos E, Johnson V, Rogers P, Suraweera N, Tomlinson I, et al. Apc Mutations in Fap-Associated Desmoid Tumours are Non-Random but Not 'Just Right'. Hum Mol Genet 2007; 16(1): 78-82.
28- Mankaney G, Leone P, Cruise M, LaGuardia L, O'Malley M, Bhatt A, et al. Gastric Cancer in Fap: A Concerning Rise in Incidence. Fam Cancer 2017; 16(3): 371-6.
29- Campos FG, Martinez CAR, Bustamante Lopez LA, Kanno DT, Nahas SC, Cecconello I. Advanced Duodenal Neoplasia and Carcinoma in Familial Adenomatous Polyposis: Outcomes of Surgical Management. J Gastrointest Oncol 2017; 8(5): 877-84.
30- Groves CJ, Saunders BP, Spigelman AD, Phillips RK. Duodenal Cancer in Patients with Familial Adenomatous Polyposis (Fap): Results of a 10 Year Prospective Study. Gut 2002; 50(5): 636-41.
31- Bülow S, Björk J, Christensen IJ, Fausa O, Järvinen H, Moesgaard F, et al. Duodenal Adenomatosis in Familial Adenomatous Polyposis. Gut 2004; 53(3): 381-6.
32- Deibert B, Ferris L, Sanchez N, Weishaar P. The Link Between Colon Cancer and Congenital Hypertrophy of the Retinal Pigment Epithelium (Chrpe). Am J Ophthalmol Case Rep 2019; 15: 100524.
33- Wijn MA, Keller JJ, Giardiello FM, Brand HS. Oral and Maxillofacial Manifestations of Familial Adenomatous Polyposis. Oral Dis 2007; 13(4): 360-5.
34- Groen EJ, Roos A, Muntinghe FL, Enting RH, de Vries J, Kleibeuker JH, et al. Extra-Intestinal Manifestations of Familial Adenomatous Polyposis. Ann Surg Oncol 2008; 15(9): 2439-50.
35- Vasen HF, Möslein G, Alonso A, Aretz S, Bernstein I, Bertario L, et al. Guidelines for the Clinical Management of Familial Adenomatous Polyposis (Fap). Gut 2008; 57(5): 704-13.
36- Abdullah Suhaimi SN, Nazri N, Nani Harlina ML, Md Isa N, Muhammad R. Familial Adenomatous Polyposis-Associated Papillary Thyroid Cancer. Malays J Med Sci 2015; 22(4): 69-72.
37- Farinella E, Soobrah R, Phillips RK, Clark SK. Familial Adenomatous Polyposis (Fap) and Gender. Does Gender Influence the Genetic Transmission of Fap? Fam Cancer 2010; 9(3): 405-6.
38- Waller A, Findeis S, Lee MJ. Familial Adenomatous Polyposis. J Pediatr Genet 2016; 5(2): 78-83.
39- Leppert M, Burt R, Hughes JP, Samowitz W, Nakamura Y, Woodward S, et al. Genetic Analysis of an Inherited Predisposition to Colon Cancer in a Family with a Variable Number of Adenomatous Polyps. N Engl J Med 1990; 322(13): 904-8.
40- Ripa R, Bisgaard ML, Bülow S, Nielsen FC. De Novo Mutations in Familial Adenomatous Polyposis (Fap). Eur J Hum Genet 2002; 10(10): 631-7.
41- Torrezan GT, da Silva FC, Santos EM, Krepischi AC, Achatz MI, Aguiar S Jr, et al. Mutational Spectrum of the Apc and Mutyh Genes and Genotype-Phenotype Correlations in Brazilian Fap, Afap, and Map Patients. Orphanet J Rare Dis 2013; 8: 54.
42- Newton KF, Mallinson EK, Bowen J, Lalloo F, Clancy T, Hill J, et al. Genotype-Phenotype Correlation in Colorectal Polyposis. Clin Genet 2012; 81(6): 521-31.
43- Nieuwenhuis MH, Vasen HF. Correlations between Mutation Site in Apc and Phenotype of Familial Adenomatous Polyposis (Fap): A Review of the Literature. Crit Rev Oncol Hematol 2007; 61(2): 153-61.
44- Dinarvand P, Davaro EP, Doan JV, Ising ME, Evans NR, Phillips NJ, et al. Familial Adenomatous Polyposis Syndrome: An Update and Review of Extraintestinal Manifestations. Arch Pathol Lab Med 2019; 143(11): 1382-98.
45- Renkonen ET, Nieminen P, Abdel-Rahman WM, Moisio AL, Järvelä I, Arte S, et al. Adenomatous Polyposis Families That Screen Apc Mutation-Negative By Conventional Methods Are Genetically Heterogeneous. J Clin Oncol 2005; 23(24): 5651-9.
46- Aihara H, Kumar N, Thompson CC. Diagnosis, Surveillance, and Treatment Strategies for Familial Adenomatous Polyposis: Rationale and Update. Eur J Gastroenterol Hepatol 2014; 26(3): 255-62.
47- Wang D, Liang S, Zhang X, Dey SK, Li Y, Xu C, et al. Targeted Next-Generation Sequencing Approach for Molecular Genetic Diagnosis of Hereditary Colorectal Cancer: Identification of a Novel Single Nucleotide Germline Insertion in Adenomatous Polyposis Coli Gene Causes Familial Adenomatous Polyposis. Mol Genet Genomic Med 2019; 7(1): e00505.
48- Herzig D, Hardiman K, Weiser M, You N, Paquette I, Feingold DL, et al. The American Society of Colon and Rectal Surgeons Clinical Practice Guidelines for the Management of Inherited Polyposis Syndromes. Dis Colon Rectum 2017; 60(9): 881-94.
49- Tajika M, Tanaka T, Oonishi S, Yamada K, Kamiya T, Mizuno N, et al. Endoscopic Management of Adenomas in the Ileal Pouch and the Rectal Remnant after Surgical Treatment in Familial Adenomatous Polyposis. J Clin Med 2022; 11(12): 3562.
50- Hyer W, Cohen S, Attard T, Vila-Miravet V, Pienar C, Auth M, et al. Management of Familial Adenomatous Polyposis in Children and Adolescents: Position Paper from the Espghan Polyposis Working Group. J Pediatr Gastroenterol Nutr 2019; 68(3): 428-41.
51- Tudyka VN, Clark SK. Surgical Treatment in Familial Adenomatous Polyposis. Ann Gastroenterol 2012; 25(3): 201-6.
52- Anele CC, Xiang J, Martin I, Hawkins M, Man R, Clark SK, et al. Regular Endoscopic Surveillance and Polypectomy is Effective in Managing Rectal Adenoma Progression Following Colectomy and Ileorectal Anastomosis in Patients with Familial Adenomatous Polyposis. Colorectal Dis 2022; 24(3): 277-83.
53- Thiruvengadam SS, Lopez R, O'Malley M, LaGuardia L, Church JM, Kalady M, et al. Spigelman Stage Iv Duodenal Polyposis Does Not Precede Most Duodenal Cancer Cases in Patients with Familial Adenomatous Polyposis. Gastrointest Endosc 2019; 89(2): 345-54. e2.
54- Lopez-Ceron M, van den Broek FJ, Mathus-Vliegen EM, Boparai KS, van Eeden S, Fockens P, et al. The Role of High-Resolution Endoscopy and Narrow-Band Imaging in the Evaluation of Upper Gi Neoplasia in Familial Adenomatous Polyposis. Gastrointest Endosc 2013; 77(4): 542-50.
55- Campos FG, Sulbaran M, Safatle-Ribeiro AV, Martinez CA. Duodenal Adenoma Surveillance in Patients with Familial Adenomatous Polyposis. World J Gastrointest Endosc 2015; 7(10): 950-9.
56- Belfiore A, Ciniselli CM, Signoroni S, Gariboldi M, Mancini A, Rivoltini L, et al. Preventive Anti-Inflammatory Diet to Reduce Gastrointestinal Inflammation in Familial Adenomatous Polyposis Patients: A Prospective Pilot Study. Cancer Prev Res (Phila) 2021; 14(10): 963-72.
57- Nieuwenhuis MH, Mathus-Vliegen EM, Baeten CG, Nagengast FM, van der Bijl J, van Dalsen AD, et al. Evaluation of Management of Desmoid Tumours Associated with Familial Adenomatous Polyposis in Dutch Patients. Br J Cancer 2011; 104(1): 37-42.
58- Iwama T, Akasu T, Utsunomiya J, Muto T. Does a Selective Cyclooxygenase-2 Inhibitor (Tiracoxib) Induce Clinically Sufficient Suppression of Adenomas in Patients with Familial Adenomatous Polyposis? A Randomized Double-Blind Placebo-Controlled Clinical Trial. Int J Clin Oncol 2006; 11(2): 133-9.
59- Burke CA, Dekker E, Lynch P, Samadder NJ, Balaguer F, Hüneburg R,, et al. Eflornithine Plus Sulindac for Prevention of Progression in Familial Adenomatous Polyposis. N Engl J Med 2020; 383(11): 1028-39.
60- Lynch PM, Burke CA, Phillips R, Morris JS, Slack R, Wang X, et al. An International Randomised Trial of Celecoxib Versus Celecoxib Plus Difluoromethylornithine in Patients with Familial Adenomatous Polyposis. Gut 2016; 65(2): 286-295.
61- Dovizio M, Tacconelli S, Ricciotti E, Bruno A, Maier TJ, Anzellotti P, et al. Effects of Celecoxib on Prostanoid Biosynthesis and Circulating Angiogenesis Proteins in Familial Adenomatous Polyposis. J Pharmacol Exp Ther 2012; 341(1): 242-50.
62- Aelvoet AS, Buttitta F, Ricciardiello L, Dekker E. Management of Familial Adenomatous Polyposis and Mutyh-Associated Polyposis; New Insights. Best Pract Res Clin Gastroenterol 2022; 58-59: 101793.
63- Kantor M, Sobrado J, Patel S, Eiseler S, Ochner C. Hereditary Colorectal Tumors: A Literature Review on Mutyh-Associated Polyposis. Gastroenterol Res Pract 2017; 2017: 8693182.
64- Rashid M, Fischer A, Wilson CH, Tiffen J, Rust AG, Stevens P, et al. Adenoma Development in Familial Adenomatous Polyposis and Mutyh‐Associated Polyposis: Somatic Landscape and Driver Genes. J Pathol 2016; 238(1): 98-108.
65- Disel U, Sivakumar S, Pham T, Fleischmann Z, Anu RI, Sokol ES, et al. Increased Kras G12c Prevalence, High Tumor Mutational Burden, and Specific Mutational Signatures are Associated with Mutyh Mutations: A Pan-Cancer Analysis. Oncologist 2023: oyad230.
66- Toboeva MK, Shelygin YA, Frolov SA, Kuzminov MA, Tsukanov AS. Mutyh-Associated Polyposis. Ter Arkh 2019; 91(2): 97-100.
67- Out AA, Tops CM, Nielsen M, Weiss MM, van Minderhout IJ, Fokkema IF, et al. Leiden Open Variation Database of the Mutyh Gene. Hum Mutat 2010; 31(11): 1205-15.
68- Moisio AL, Järvinen H, Peltomäki P. Genetic and Clinical Characterisation of Familial Adenomatous Polyposis: A Population Based Study. Gut 2002; 50(6): 845-50.
69- Knudsen A, Bülow S, Tomlinson I, Möslein G, Heinimann K, Christensen I, et al. Attenuated Familial Adenomatous Polyposis: Results from an International Collaborative Study. Colorectal Dis 2010; 12(10Online): e243-9.
70- Grover S, Kastrinos F, Steyerberg EW, Cook EF, Dewanwala A, Burbidge LA, et al. Prevalence and Phenotypes of Apc and Mutyh Mutations in Patients with Multiple Colorectal Adenomas. Jama 2012; 308(5): 485-92.
71- Guarinos C, Juárez M, Egoavil C, Rodríguez-Soler M, Pérez-Carbonell L, Salas R, et al. Prevalence and Characteristics of Mutyh-Associated Polyposis in Patients with Multiple Adenomatous and Serrated Polyps. Clin Cancer Res 2014; 20(5): 1158-68.
72- Seguí N, Navarro M, Pineda M, Köger N, Bellido F, González S, et al. Exome Sequencing Identifies Mutyh Mutations in a Family with Colorectal Cancer and an Atypical Phenotype. Gut 2015; 64(2): 355-6.
73- Morak M, Heidenreich B, Keller G, Hampel H, Laner A, de la Chapelle A, et al. Biallelic Mutyh Mutations Can Mimic Lynch Syndrome. Eur J Hum Genet 2014; 22(11): 1334-7.
74- Win AK, Hopper JL, Jenkins MA. Association between Monoallelic Mutyh Mutation and Colorectal Cancer Risk: A Meta-Regression Analysis. Fam Cancer 2011; 10(1): 1-9.
75- Theodoratou E, Campbell H, Tenesa A, Houlston R, Webb E, Lubbe S, et al. A Large-Scale Meta-Analysis to Refine Colorectal Cancer Risk Estimates Associated with Mutyh Variants. Br J Cancer 2010; 103(12): 1875-84.
76- Vogt S, Jones N, Christian D, Engel C, Nielsen M, Kaufmann A, et al. Expanded Extracolonic Tumor Spectrum in Mutyh-Associated Polyposis. Gastroenterology 2009; 137(6): 1976-85. e1-10
77- Nielsen M, Morreau H, Vasen HF, Hes FJ. Mutyh-Associated Polyposis (Map). Crit Rev Oncol Hematol 2011; 79 (1): 1-16.
78- Pitroski CE, Cossio SL, Koehler-Santos P, Graudenz M, Prolla JC, Ashton-Prolla P. Frequency of the Common Germline Mutyh Mutations P. G396d and P. Y179c in Patients Diagnosed with Colorectal Cancer in Southern Brazil. Int J Colorectal Dis 2011; 26(7): 841-6.
ارسال پیام به نویسنده مسئول
| بازنشر اطلاعات | |
 |
این مقاله تحت شرایط Creative Commons Attribution-NonCommercial 4.0 International License قابل بازنشر است. |
