

دوره 31، شماره 1 - ( فروردین 1402 )
جلد 31 شماره 1 صفحات 6300-6286 |
برگشت به فهرست نسخه ها


![]()
![]()
![]()
Download citation:
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:



BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
Molayi-Asl A, Sepehri S. Identification and Introduction of Possible Inhibitors of Plasmodium Falciparum Lactate Dehydrogenase Enzyme using Computational Techniques of Drug Design and Virtual Screening based on Macromolecules. JSSU 2023; 31 (1) :6286-6300
URL: http://jssu.ssu.ac.ir/article-1-5844-fa.html
URL: http://jssu.ssu.ac.ir/article-1-5844-fa.html
مولایی اصل اسما، سپهری ساقی. شناسایی و معرفی مهارکنندههای احتمالی آنزیم لاکتات دهیدروژناز پلاسمودیوم فالسیپاروم با استفاده از تکنیکهای محاسباتی طراحی دارو و غربالگری مجازی براساس ماکرومولکول. مجله علمي پژوهشي دانشگاه علوم پزشكي شهید صدوقی يزد. 1402; 31 (1) :6286-6300



متن کامل [PDF 1248 kb]
(728 دریافت)
| چکیده (HTML) (1791 مشاهده)
References:
1- World Malaria Report. 2020. Available at: https://www.who.int/publications/i/item/9789240015791. Accessed March 2021.
2- Muhseen ZT, Hameed AR, Al-Bhadly O, Ahmad S, Li G. Natural Products for Treatment of Plasmodium Falciparum Malaria: An Integrated Computational Approach. Comput Biol Med 2021; 134: 104415.
3- Varela-Aramburu S, Ghosh C, Goerdeler F, Priegue P, Moscovitz O, Seeberger PH. Targeting and Inhibiting Plasmodium Falciparum Using Ultra-Small Gold Nanoparticles. ACS Appl Mater Interfaces 2020; 12(39): 43380-87.
4- Flegg JA, Metcalf CJE, Gharbi M, Venkatesan M, Shewchuk T, Sibley CH, Guerin PJ. Trends in Antimalarial Drug Use in Africa. Am J Trop Med 2013; 89(5): 857-65.
5- Waingeh, VF, Groves AT, Eberle JA. Binding of Quinoline-Based Inhibitors to Plasmodium falciparum Lactate Dehydrogenase: A Molecular Docking Study. Open J Biophys 2013; 3(4): 285-90.
6- Penna-Coutinho J, Cortopassi WA, Oliveira AA, França TCC, Krettli AU. Antimalarial Activity of Potential Inhibitors of Plasmodium falciparum Lactate Dehydrogenase Enzyme Selected by Docking Studies. PLoS ONE 2011; 6(7): e21237.
7- Singh R, Bhardwaj V, Purohit R. Identification of a Novel Binding Mechanism of Quinoline Based Molecules with Lactate Dehydrogenase of Plasmodium Falciparum. J Biomol Struct Dyn 2021; 39(1): 348-56.
8- Reynolds CR, Muggleton SH, Sternberg MJE. Incorporating Virtual Reactions into a Logic-based Ligand-based Virtual Screening Method to Discover New Leads. Mol Inform 2015; 34(9): 615-25.
9- Sepehri S, Saghaie L, Fassihi A. Anti-HIV-1 Activity Prediction of Novel Gp41 Inhibitors Using Structure-Based Virtual Screening and Molecular Dynamics Simulation. Mol Inform 2017; 36(3): 1600060.
10- Insuasty B, Ramírez J, Becerra D, Echeverry C, Quiroga J, Abonia R, et al. An Efficient Synthesis of New Caffeine-Based Chalcones, Pyrazolines and Pyrazolo[3,4-b][1,4]diazepines as Potential Antimalarial, Antitrypanosomal and Antileishmanial Agents. Eur J Med Chem 2015; 93: 401-13.
11- Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. Automated Docking Using a Lamarckian Genetic Algorithm and an Empirical Binding Free Energy Function. J Comput Chem 1998; 19(14): 1639-62.
12- Mills N. ChemDraw Ultra 10.0 CambridgeSoft, 100 CambridgePark Drive, Cambridge, MA 02140. Www.cambridgesoft.com. Commercial Price: $1910 for download, $2150 for CD-ROM; Academic Price: $710 for download, $800 for CD-ROM. J Am Chem Soc 2006; 128(41): 13649-50.
13- Jeddi B, Saberi S, Menéndez JC, Sepehri S. Synthesis and Biological Evaluation of Tetrahydropyrimidine and Dihydropyridine Derivatives against Leishmania Major. Acta Parasitol 2022; 67(1): 255-66.
14- Saddala MS, Kumar KK, Rani AU. In Silico Inhibitors for Plasmodium Falciparum Lactate Dehydrogenase. J Bioinforma 2014; 14(2): 146-59.
15- Mishra M, Agarwal S, Dixit A, Mishra VK, Kashaw V, Agrawal RK, et al. Integrated Computational Investigation to Develop Molecular Design of Quinazoline Scaffold as Promising Inhibitors of Plasmodium Lactate Dehydrogenase. J Mol Str 2020; 1207: 127808.
16- Kaushik D, Paliwal D, Kumar A. 2D QSAR and Molecular Docking Studies of Chloroquine Thiazolidinone Derivatives as Potential pfLDH Inhibitors of Plasmodium Falciparum. Int J Pharmacol Pharm Sci 2015; 2(5): 42-53.
17- Shadrack DM, Nyandoro SS, Munissi JJE, Mubofu EB. In Silico Evaluation of Anti-malarial Agents from Hoslundia Opposita as Inhibitors of Plasmodium Falciparum Lactate Dehydrogenase (PfLDH) Enzyme. Comput Mol Biosci 2016; 6(2): 23-32.
18- Zakaria NH, WAI L, Hassan NI. Molecular Docking Study of the Interactions between Plasmodium Falciparum Lactate Dehydrogenase and 4-Aminoquinoline Hybrids. Sains Malays 2020; 49(8): 1905-13.
19- Chaniad P, Mungthin M, Payaka A, Viriyavejakul P, Punsawad C. Antimalarial Properties and Molecular Docking Analysis of Compounds From Dioscorea Bulbifera L. as New Antimalarial Agent Candidates. BMC Complement Med Ther 2021; 21: 144.
20- Shamsuddin MA, Ali AH, Zakaria NH, Mohammat MF, Hamzah AS, Shaameri Z, et al. Synthesis, Molecular Docking, and Antimalarial Activity of Hybrid 4-Aminoquinoline-pyrano[2,3-c]pyrazole Derivatives. Pharmaceuticals 2021; 14(11): 1174.
21- Oluyemi WM, Samuel BB, Adewumi AT, Adekunle YA, Soliman MES, Krenn L. An Allosteric Inhibitory Potential of Triterpenes from Combretum racemosum on the Structural and Functional Dynamics of Plasmodium falciparum Lactate Dehydrogenase Binding Landscape. Chem Biodivers 2022; 19(2): e202100646.
متن کامل: (1046 مشاهده)
مقدمه
مالاریا کشندهترین بیماری عفونی است که بیش از 300 سال قبل آغاز گردیده اما طبق گزارش سازمان بهداشت جهانی طی دو دهه اخیر، کاهش چشمگیر موارد مرگ و میر از این بیماری مشاهده شده است. در سال 2019، 229 میلیون نفر در 87 کشور به مالاریا مبتلا و تعداد کل 409000 مرگ و میر در سراسر جهان گزارش شد (1). مالاریا توسط انگلهای تک یاختهای جنس پلاسمودیوم ایجاد و از طریق پشه آنوفل منتشر میگردد. پنج گونه شناخته شده مالاریا شامل پلاسمودیوم مالاریا، پلاسمودیوم اوالا، پلاسمودیوم ویواکس، پلاسمودیوم ناولسکی و پلاسمودیوم فالسیپاروم وجود دارند. از بین پنج گونه انگلی پلاسمودیوم، پلاسمودیوم فالسیپاروم انسان را آلوده میکند و از میان همه آنها کشندهتر است (2). در غیاب یک واکسن موثر، استفاده از روشهای شیمیدرمانی به عنوان عوامل ضد مالاریا تنها راه باقیمانده برای مدیریت و پیشگیری از بیماری مالاریا است. تحقیقات نشان دادند که اثربخشی بیشتر ترکیبات ضد مالاریا به گونههای پلاسمودیوم مقاومت نشان میدهند. مقاومت در برابر تمام گروههای اصلی داروها شامل 4-آمینوکوئینولینها (کلروکین، آمودیاکین و پیپراکین)، آنتی فولاتها، آریل آمینو الکلها (کینین، لومفانترین و مفلوکین)، مشتقات آرتمیزینین، آنتیبیوتیکها (کلیندامایسین و داکسی سایکلین) و نپتوکوکینون گزارش شده است (3). این امر باعث میشود محققان بر روی اهداف پلاسمودیوم فالسیپاروم (pf) کار کنند تا داروهای موثر بر ضد آن و سایر گونههای مالاریا را که بر انسان موثر است معرفی نمایند. کلروکین بهطور وسیعی از دهه 1940 به عنوان داروی انتخابی با هزینه قابل قبول برای درمان همه انواع مالاریا استفاده شده است. ویژگیهای مناسب کلروکین باعث استفاده گسترده از آن و نهایتاً مقاومت به آن در pf منجر شد. در نتیجه این گونه انگل مسئول گسترده ترین مشکلات مالاریای انسانی گردید (4). این امر نیاز به توسعه داروهای ضد مالاریا جدید را افزایش داده است که در کنترل مالاریا موثرتر باشد. اهداف مختلفی برای داروهای ضد مالاریا مانند پلاسمپسین I، II و V، فالسیپایین2، لاکتات دهیدروژناز pf و پیریدوکسال کیناز وجود دارد. از میان این اهداف، آنزیم لاکتات دهیدروژناز (LDH) یک هدف ارجح برای داروهای ضد مالاریا است؛ زیرا تولید آدنوزین تری فسفات (ATP) در پلاسمودیوم را از طریق مسیر گلیکولیتیک کنترل میکند. مسیرهای گلیکولیتیک و آنزیمهای مرتبط به دلیل وابستگی انگلی آنها به چرخه گلیکولیز برای تولید انرژی، اهداف دارویی حیاتی هستند (5). pfLDH یک نقش مهم در آخرین مرحله گلیکولیز دارد و تبدیل پیروات به لاکتات را کاتالیز میکند. آنزیم pfLDH بیشتر با توسعه NAD+ مرتبط است که برای آنزیم گلیکولیتیک گلیسرآلدئید-3-فسفات دهیدروژناز مورد نیاز است (6). انگلهای pf برای تولید انرژی خود به این آنزیم نیازمند هستند که برای عملکرد، رشد و توسعه بیوشیمیایی مهم است. بنابراین، این آنزیم یک هدف دارویی مهم در درمان مالاریا است و مهار آن باعث مرگ انگل میگردد. توالی اسیدهای آمینه برای LDH انسانی و pfLDH شباهت کمی وجود دارد (7). بنابراین، هدفگیری انتخابی این آنزیم گلیکولیتیک در pfLDH قابلتوجه است اما LDH انسانی را متوقف نمیکند. این ویژگیها، pfLDH را به عنوان یک هدف مناسب برای طراحی مبتنی بر ساختار ضد مالاریاهای جدید توصیه میکند. غربالگری مجازی (VS) یک استراتژی کاربردی است که برای تمایز مولکولها براساس ویژگی مورد نظر استفاده میشود و میتواند برای شناسایی ترکیبات الگوی جدید مفید باشد. میتوان آن را به دو دسته گسترده تقسیم کرد: مبتنی بر ساختار و مبتنی بر لیگاند (8). روشهای غربالگری مجازی مبتنی بر ساختار یا گیرنده (SBVS) زمانی بسیار مناسب هستند که اطلاعات مربوط به ساختار هدف در دسترس باشد. در اینروش حالتهای اتصال برای هر لیگاند پیشبینی میشود. رویکردهای غربالگری مجازی مبتنی بر لیگاند (LBVS) از اطلاعات دادههای ساختار-فعالیت مولکولهای فعال شناساییشده با هدف شناسایی ترکیبات ساختاری متنوع با زیست فعالیتی مشابه استفاده میکنند. ترکیبات الگوی شناسایی شده از VS، به عنوان نقطه شروعی برای بهینهسازی ترکیبات الگو در برنامههای کشف دارو میباشد (9). در این مطالعه، به منظور شناسایی و معرفی مهارکنندههای بالقوه جهت مهار آنزیم pfLDH با استفاده از روشهای غربالگری مجازی مبتنی بر ساختار، مهارکنندههای احتمالی آنزیم pfLDH به عنوان ترکیبات دارویی ضد مالاریا طراحی شدند. بدین منظور کتابخانهای از ترکیبات تشکیل شد و با بهرهگیری از فیلتراسیونهای مختلف ترکیبات مورد ارزیابی قرار گرفتند. فلوچارت فرایند VS در شکل 1 نشان داده شده است.

شکل 1: فلوچارت فرایند کلی غربالگری مجازی
روش بررسی
این پژوهش به صورت توصیفی-تحلیلی صورت گرفت. در این مطالعه ترکیب 7-((4-(6،2-دی اکسو-7،3،1-تری متیل-7،6،3،2- تتراهیدرو-H1-پورین-8-یل)اکسی)-3-متوکسی فنیل)-3-متیل-1-فنیل-5-(5،4،3-تری متوکسی فنیل)-8،7،6،1-تتراهیدروپیرازولو [4،3-b][4،1] دیازپین (ترکیب 1، شکل 2) با فعالیت ضد مالاریای قوی (مقدار IC50 برابر 3/11 ماکروگرم بر میلیلیتر و درصد مهار برابر 94/2%) که در مطالعات قبلی گزارش شده بود (10) به عنوان ترکیب الگو برای تهیه کتابخانه ترکیبات مورد استفاده قرار گرفت.
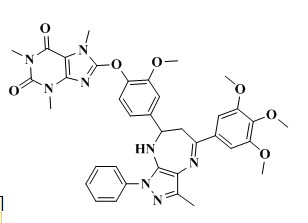
شکل 2. ساختار شیمیایی ترکیب 1
تهیه کتابخانه داده و غربالگری مجازی: قبل از شروع غربالگری مجازی، ابتدا لازم است کتابخانهای از ترکیبات تهیه شود. در این پژوهش ابتدا کتابخانه ترکیبات از پایگاه داده PubChem استخراج گردید (https://pubchem.ncbi.nlm.nih.gov). پایگاه PubChem یک پایگاه رایگان با بیش از 100 میلیون ساختار است. کتابخانه شامل 8733 ترکیب از پایگاه داده PubChem تشکیل شد. همه ساختارها در فورمت SDF ذخیره و سپس به فورمت pdbqt توسط نرمافزار PyRx 0/8 (https://pyrx.sourceforge.io) تبدیل شدند. در مرحله بعد، ترکیبات کتابخانه داخل جایگاه فعال آنزیم pfLDH با استفاده از اتوداک وینا در نرمافزار PyRx 0/8 داک گردیدند. تعداد runها برای هر آنالیز داکینگ روی 100 تنظیم شد. الگوریتم بهکار برده شده لامارکین ژنتیک بود. نتایج داکینگ براساس انرژی آزاد اتصال مرتب و ترکیبات با بالاترین انرژی آزاد اتصال انتخاب گردیدند.
بررسی خواص داروهمانندی: از میان کل ترکیبات کتابخانه، 1001 ترکیب از مرحله قبل با بالاترین انرژی آزاد اتصال انتخاب شدند و برای ارزیابی خواص داروهمانندی و پارامترهای قانون پنج لیپنیسکی وارد این مرحله گردیدند. پارامترهای مورد ارزیابی شامل وزن مولکولی، تعداد اتمهای دهنده هیدروژن، تعداد اتمهای گیرنده هیدروژن، لیپوفیلیسیته (LogP)، توپولوژی مساحت سطح قطبی (TPSA) و تعداد پیوندهای قابل چرخش است. تمام این خواص با استفاده از وب سرور Molinspiration محاسبه شد (https://www.molinspiration.com).
بررسی ویژگیهای فارماکوکینتیکی ترکیبات (ADMET): خواص فارماکوکینتیک شناخته شده تحت عنوان ADMET (جذب، پخش، متابولیسم، دفع و سمیت) موقعیت ترکیبات دارویی را در ارگانیسم بررسی میکنند. یک داروی موفق تنها منوط به داشتن قدرت اثر خوب نیست بلکه باید ویژگیهای ADMET قابل قبولی داشته باشد. بنابراین در این مرحله خواص ADMET 281 ترکیب راه یافته از مرحله قبل توسط وب سرورهای swiss ADME (http://www.swissadme.ch) و admet SAR (http://lmmd.ecust.edu.cn/admetsar2) مورد بررسی قرار گرفت. در نهایت از 281 ترکیب وارد شده 13 ترکیب توانستند پروفایل فارماکوکینتیک خوبی را نشان دهند و به مرحله بعدی راه پیدا کنند.
مطالعه شبیه سازی داکینگ مولکولی: برای انجام روش داکینگ مولکولی از نرمافزار اتوداک 4.2 استفاده شد (11). بدین منظور نرمافزار اتوداک بر روی کامپیوتر 8 هستهای تحت سیستم عامل ویندوز نصب گردید. ژنتیک الگوریتم (GA) به عنوان الگوریتم جستجوگر توسط نرمافزار مورد استفاده قرار گرفت. برنامه گرافیکی اتوداک تولز 6.5.1 (ADT) برای تهیه، انجام و آنالیز شبیهسازیهای داکینگ مولکولی بهکار رفت. در ابتدا، ساختارهای دوبعدی ترکیبات بهوسیله برنامه ChemDraw Ultra 10.0 ترسیم (12) و سپس با استفاده از نرمافزار Hyperchem8 HyperChem, Release 8.0 for Windows, Molecular Modeling System: HyperCube, 2007)) در میدان نیروی مکانیک مولکولی (MM+) و روش نیمه تجربی PM3 و الگوریتم Polak-Ribiere از نظر انرژی بهینه شدند. پس از بهینهسازی انرژی لیگاند، با استفاده از نرمافزار اتوداک تولز اتمهای هیدروژن به ساختار مولکول افزوده شدند. در مرحله بعد، اتمهای هیدروژن غیرقطبی در اتم کربن مربوطه ادغام و بار الکتریکی گستیگر (بارهای الکتریکی اتم که بهصورت تجربی محاسبه میگردد) و تعداد درجات آزادی زوایای پیچشی لیگاند با استفاده از نرمافزار اتوداک تولز محاسبه گردید. در نهایت فایل لیگاند به صورت pdbqt ذخیره شد. ساختار کریستالی سه بعدی آنزیم pfLDH از پایگاه بانک دادههای پروتئین دانلود گردید (کد شناسایی 1CET) (https://www.rcsb.org). روش انجام داکینگ و آنالیز کانفورمرها براساس توضیحات در مقالات قبلی صورت گرفت (13). در ابتدا با استفاده از نرمافزار Notpat++ و یا Discovery Studio Viewer lite 4.0 مولکولهای آب از ساختار کریستالوگرافی حذف گردیدند. سپس با استفاده از نرمافزار اتوداک تولز اتمهای هیدروژن به ساختار کریستالوگرافی افزوده شدند. در مرحله بعد، اتمهای هیدروژن غیر قطبی در اتم کربن مربوطه ادغام شده و بار الکتریکی کلمن و پارامترهای حلال پوشی ماکرومولکول محاسبه و در نهایت به صورت pdbqt ذخیره گردید. پس از تهیه فایلهای ورودی مورد نیاز داکینگ (ماکرومولکول، لیگاند و نقشه اتصال)، مطالعات داکینگ به منظور مدل سازی برهمکنشهای لیگاند-گیرنده، با استفاده از الگوریتمی تحت عنوان ژنتیک لامارکین انجام شد. در مرحله بعد، براساس حجم مولکولی لیگاندهای طراحی شده، شبکهای با ابعاد 60 × 60 × 60 آنگستروم در راستای محورهای سه گانه مختصات و فاصله نقاط شبکه 0/573 آنگستروم (یک چهارم طول پیوند ساده کربن-کربن) که در برگیرنده جایگاه فعالگیرنده بود، در نظر گرفته شد. فایل شبکه به صورت gpf ذخیره گردید. پارامترهای ذخیره شده در فایل gpf در اختیار محاسبات اتوگرید قرار گرفته است. پس از انجام عملیات داکینگ، نتایج شامل کانفورماسیونهای مولکول، انواع برهمکنشهای مولکول با پروتئین شامل برهمکنشهای هیدروژنی، هیدروفوبی و π-π با اسیدهای آمینه موجود در جایگاه اتصال پروتئینها قابل مشاهده و تجزیه و تحلیل میباشند. به منظور دستیابی به این اطلاعات از نرمافزارهای اتوداک تولز و Discovery Studio Viewer lite 4.0 استفاده شد (Accelryslnc, San Diego, CA, USA).
تجزیه و تحلیل آماری
در این مطالعه ترکیبات با پتاسیل مهار آنزیم pfLDH براساس روش غربالگری مجازی مبتنی بر ساختار شناسایی و معرفی گردید.
ملاحظات اخلاقی
کد اخلاق مصوبه این پروژه توسط دانشگاه علوم پزشکی اردبیل IR.ARUMS.REC.1397.138 است.
نتایج
تشکیل کتابخانه و غربالگری مجازی: در زمینه کشف و طراحی ترکیبات دارویی، VS میتواند به طور موثری برای انتخاب مولکولهای زیست فعال بالقوه از کتابخانه ترکیبات برای اتصال به پروتئین هدف استفاده شود. در این پروژه از پایگاه داده PubChem برای جستجوی ترکیبات با شباهت 70% به ترکیب 1 استفاده شد. نتیجه این جستجو براساس شباهت ساختاری، منجر به تشکیل یک کتابخانه با 8733 ترکیب گردید. سپس VS بر روی ترکیبات جمعآوری شده توسط نرمافزار PyRx انجام شد. بعد از اتمام کار 1001 ترکیب با انرژی آزاد اتصال برابر و بالاتر از Kcal/mol 8/79- برای مرحله بعد انتخاب شدند (برابر و بالاتر از انرژی آزاد اتصال ترکیب 1، Kcal/mol 79/8-).
خواص داروهمانندی: یکی از فاکتورهای مهم برای کشف و پیشرفت ترکیبات زیست فعال به عنوان یک داروی خوراکی فراهمی زیست بالای آنها است. برای پیش بینی ترکیبات زیست فعال به عنوان یک داروی خوراکی خوب باید معیارهای زیر را مورد توجه قرار داد: پیوندهای قابل چرخش مولکول که تحت عنوان انعطافپذیری مولکول شناخته میشود، جذب گوارشی خوب و TPSA پایین (مجموع پیوندهای هیدروژنیدهنده و پذیرنده). علاوه بر آنها، قانون پنج لیپنسکی ویژگیهای داروهمانندی شامل جرم مولکولی، مقادیر پیوندهای هیدروژنی دهنده و پذیرنده و LogP را معرفی میکند. مجموع این معیارها کمک میکند که ترکیباتی با خصوصیات فارماکوکینتیک بهتر در بدن انسان برای تجویز خوراکی معرفی گردد. در این فاز، پارامترهای فیزیکوشیمیایی مانند CLogP، TPSA، جرم مولکولی، پیوندهای هیدروژنی دهنده و گیرنده، و پیوندهای قابل چرخش بر روی ترکیبات انتخاب شده از مرحله قبل پیادهسازی شد. نهایتاً، 281 ترکیب از معیارهای ذکر شده تبعیت کردند و خواص داروهمانندی مناسبی را نشان دادند.
خواص فارماکوکینتیک (ADMET): محاسبه خواص فارماکوکینتیک ترکیبات در شناسایی اولیه آنها به عنوان ترکیبات الگو قابلتوجه میباشد. بررسی این خواص منجر به حذف کاندیدهای دارویی ضعیف میشود. در این مرحله خواص فیزیکوشیمیایی و فارماکوکینتیکی ترکیبات انتخاب شده توسط وب سرورهای swissADME و admetSAR بررسی گردید. پارامترهای مورد بررسی شامل مقدار لیپوفیلیسیتی، نفوذپذیری سد خونی-مغزی (BBB)، حلالیت، جذب و متابولیسم و همچنین مهار آنزیمهای کبدی و p-گلیکوپروتئین میباشد. در این مرحله، توسط وب سرورهای swissADME و admetSAR خواص ADMET 281 ترکیب منتخب از مرحله قبل مورد بررسی قرار گرفت. از این تعداد، 13 ترکیب با بهترین خواص فارماکوکینتیک انتخاب شدند. نتایج خواص دارو همانندی و فارماکوکینتیک 13 ترکیب منتخب در جداول 1 و 2 نشان داده شده است.
جدول 1: خواص داروهمانندی ترکیبات منتخب حاصل از غربالگری مجازی
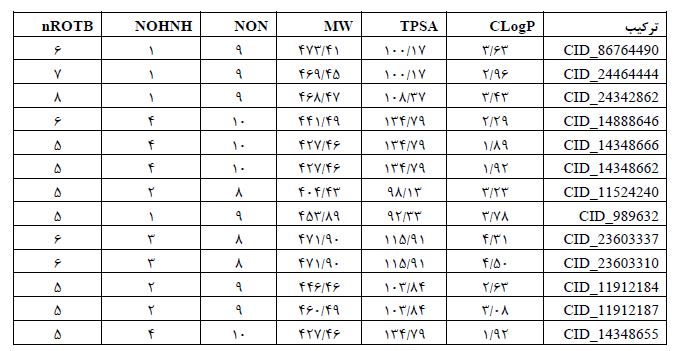
TPSA: سطح توپولوژیک قطبی؛ MW: جرم مولکولی؛ NON: تعداد پیوندهای هیدروژنی پذیرنده؛ NOHNH: تعداد پیوندهای هیدروژنی دهنده؛ nROTB: تعداد پیوندهای قابل چرخش
جدول 2. ویژگیهای فارماکوکینتیکی ترکیبات منتخب حاصل از غربالگری مجازی
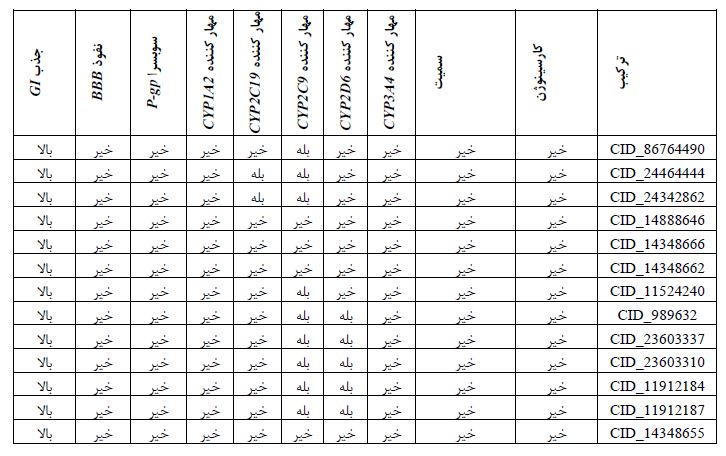
GI: جذب گوارشی؛ BBB: سد خونی-مغزی؛ P-gp: P-گلیکوپروتئین
مطالعات شبیهسازی داکینگ مولکولی: شبیهسازی داکینگ مولکولی برای مطالعه حالتهای اتصال ترکیبات منتخب داخل جایگاه فعال آنزیم انجام شد. در ابتدا به منظور اعتبار سنجی پروتکل داکینگ، لیگاند کلروکین (کوکریستال) از جایگاه فعال آنزیم حذف شد و مجدداً در جایگاه فعال داک گردید. تجزیه و تحلیل نتایج تحت عنوان ریشه انحراف میانگین مربع (RMSD) بین حالتهای داک شده و کریستالوگرافی شده انجام گردید. مقدار RMSD بهدست آمده بین کانفورماسیون داک شده کلروکین و حالت کریستال شده برابر 1/03 آنگستروم بود. براساس شاخص بینالمللی مقادیر زیر 2 آنگستروم مورد پذیرش میباشد. بعد از اعتبارسنجی پروتکل داکینگ، بررسی نتایج داکینگ 13 ترکیب منتخب از مرحله قبل نشان داد که تمام این ترکیبات فضای مشابه را درون جایگاه فعال آنزیم اشغال میکنند. ساختار شیمیایی این 13 ترکیب در شکل 3 نشان داده شده است.
داکینگ مولکولی 13 ترکیب حاصل از غربالگری مجازی در جایگاه فعال آنزیم pfLDH با کد شناسایی 1CET در نرمافزار داکینگ انجام گردید (جدول 3).
بالاترین انرژی آزاد اتصال ( Kcal/mol29/10-) برای برهمکنش CID_23603310 با اسید آمینههای جایگاه فعال به صورت زیر تفسیر میشود (شکل 4): دو پیوند هیدروژنی بین اتم نیتروژن حلقه پیریمیدین و NH آمیدی ترکیب و اسید آمینه Gly99 تشکیل شد. برهمکنشهای هیدروفوب لیگاند با اسید آمینههای Met30، Ile31، Gly27، Ile119، Asp53، Phe52، Thr101، Phe100، Ala98، Thr97 و Gly99 مشاهده گردید. ترکیب CID_23603337 با انرژی آزاد اتصال 9/06- کیلوکالری بر مول برهمکنشهای زیر را نشان داد (شکل 5): یک پیوند هیدروژنی بین اتم نیتروژن حلقه پیریمیدین و اسید آمینه Gly99 تشکیل شد. اسیدآمینههای Phe100، Thr97، Ala98، Val138، Val26، Phe52، Ile119، Tyr85 و Ile31 با ترکیب CID_23603337 برهمکنشهای هیدروفوب تشکیل دادند. حالت اتصال CID_11912187 در جایگاه فعال آنزیم در شکل 6 مشاهده میشود. این ترکیب دو پیوند هیدروژنی نشان داد؛ دو پیوند هیدروژنی بین NH گروه آمیدی متصل به حلقه فنیل و NH متصل به حلقه پیریمیدین و اسید آمینه Glu122. این ترکیب همچنین برهمکنشهای هیدروفوب با اسید آمینه های Ile121، Ile119، Phe52، Ile54، Ala98، Gly99 و Phe100 تشکیل داد. ترکیب CID_11912184 با انرژی آزاد اتصال 9/00- کیلوکالری بر مول دو پیوند هیدروژنی تشکیل دادند (شکل 7): یک پیوند هیدروژنی بین اتم اکسیژن بخش دی اکسولون با اسیدآمینه Tyr85 و یک پیوند هیدروژنی بین NH متصل به حلقه پیریمیدین با اسید آمینه Glu122. اسید آمینههای Ile54، His126، Ile119، Val26، Phe52، Gly27، Ala98 و Asp53 با ترکیب برهمکنش هیدروفوب تشکیل دادند.
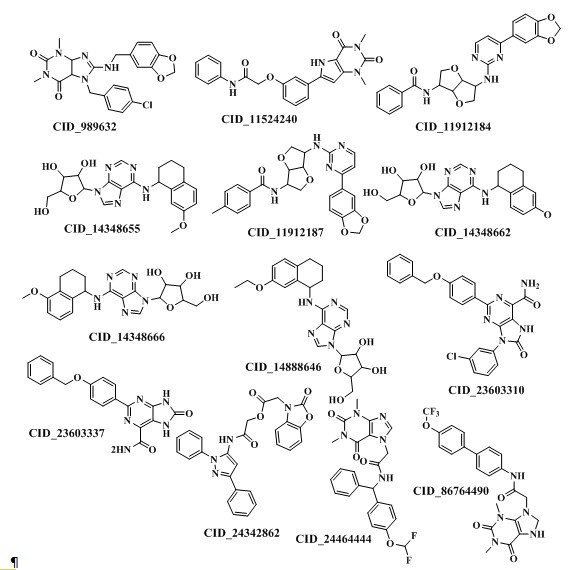
شکل 3: ساختارهای شیمیایی 13 ترکیب منتخب راه یافته به مرحله داکینگ مولکولی
جدول 3: انرژی آزاد اتصال و برهمکنشهای هیدروژنی، هیدروفوبی و π-π ترکیبات منتخب حاصل از غربالگری مجازی در جایگاه فعال آنزیم pfLDH

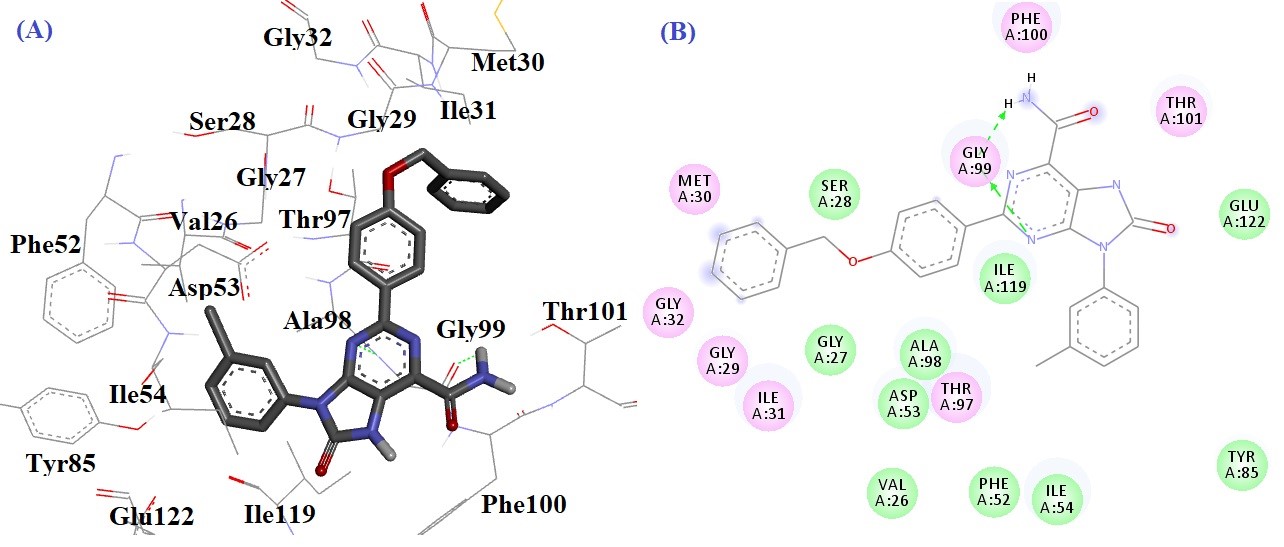
شکل 4: حالت اتصال و برهمکنشهای (A) سه بعدی (B) دو بعدی ترکیب CID_23603310 در جایگاه فعال آنزیم pfLDH
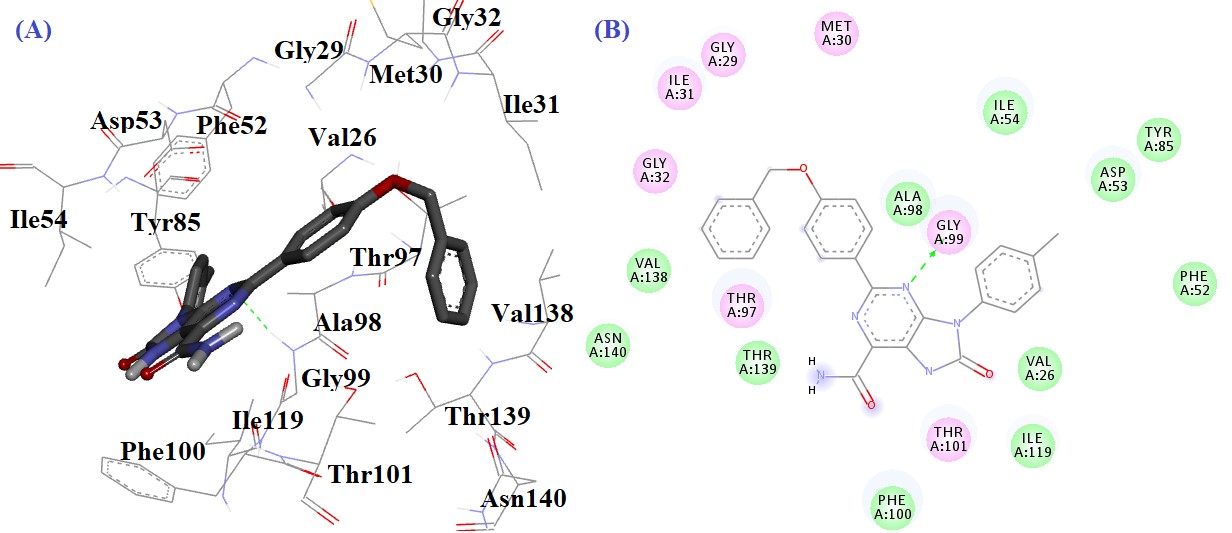
شکل 5: حالت اتصال و برهمکنشهای (A) سه بعدی (B) دو بعدی ترکیب CID_23603337 در جایگاه فعال آنزیم pfLDH
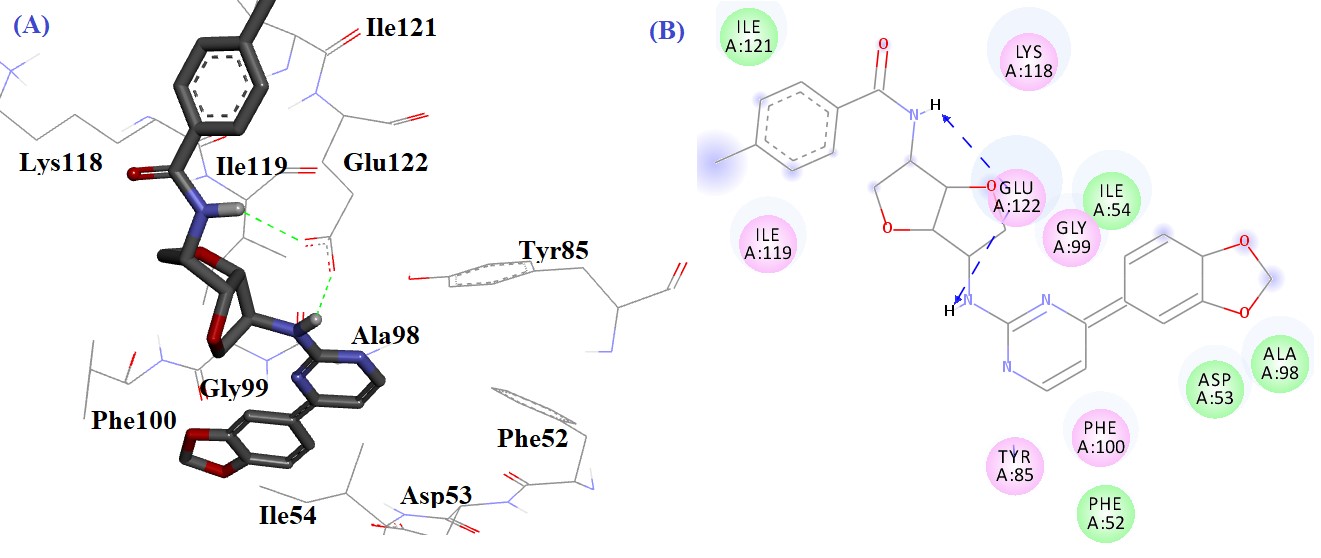
شکل 6: حالت اتصال و برهمکنشهای (A) سه بعدی (B) دو بعدی ترکیب CID_11912187 در جایگاه فعال آنزیم pfLDH
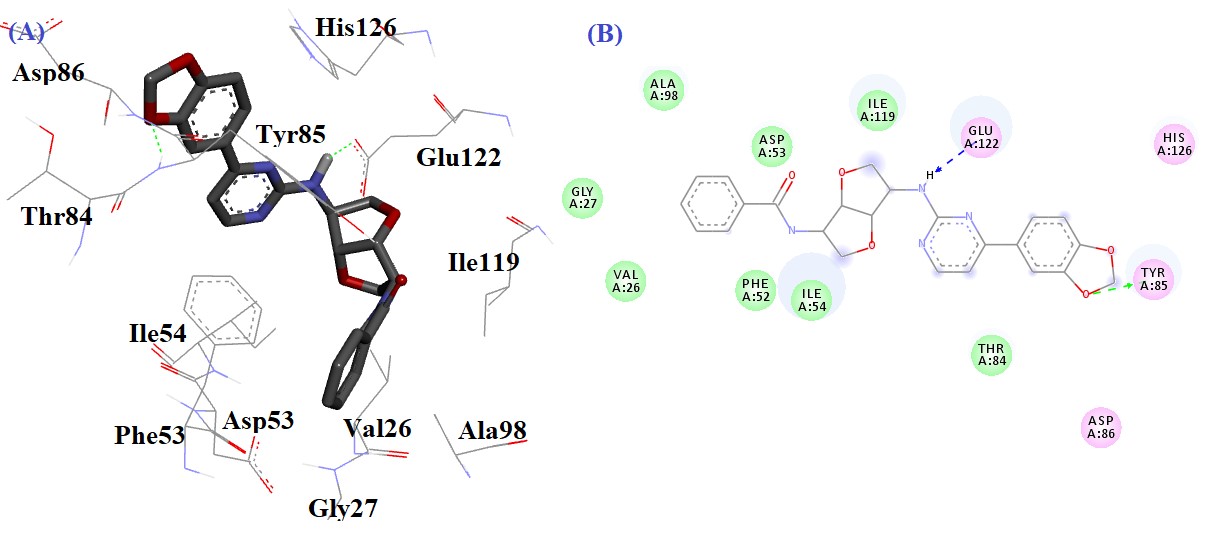
شکل 7: حالت اتصال و برهمکنشهای (A) سه بعدی (B) دو بعدی ترکیب CID_11912184 در جایگاه فعال آنزیم pfLDH
بحث
دو معیار مهم در تعیین بهترین حالت داک شده، بیشترین (منفیترین) انرژی آزاد اتصال تخمین زده شده و همچنین بیشترین برهمکنشهای مناسب با اسید آمینههای اصلی جایگاه فعال آنزیم pfLDH میباشند. 13 ترکیب نهایی با خصوصیات داروهمانندی و فارماکوکینتیک مناسب به مرحله شبیهسازی داکینگ مولکولی راه یافتند. طبق نتایج حاصل از داکینگ مولکولی همه چهار ترکیب مذکور انرژی آزاد اتصال منفیتری نسبت به کلروکین نشان داده اند. ساختار دو ترکیب CID_11912184 و CID_11912187 مشابه یکدیگر است. ساختار CID_11912187 یک گروه متیل اضافه نسبت به CID_11912184 دارد. این گروه متیل لیپوفیل به نظر میآید تا حدی اثر مثبت روی اتصال ترکیب در جایگاه فعال آنزیم داشته باشد. هر دو این ترکیبات انرژی آزاد اتصال بهتری نسبت به کلروکین و ترکیب 1 نشان دادند. ترکیب CID_11912184 یک پیوند هیدروژنی و ترکیب CID_1912187 دو پیوند هیدروژنی با اسیدآمینه Glu122 تشکیل میدهند. در ترکیب CID_11912187 گروه NH متصل به حلقه پیریمیدینی و آمیدی هر دو با Glu122 پیوند هیدروژنی تشکیل میدهند در حالیکه ترکیب CID_11912184 تنها NH متصل به حلقه پیریمیدینی با این اسید آمینه پیوند هیدروژنی تشکیل میدهد. علاوه بر آن این ترکیب از سر اکسیژن بنزودی اکسول با اسید آمینه Tyr85 پیوند هیدروژنی تشکیل میدهد. با توجه به نتایج مشاهده شده و مطالعات قبلی تشکیل پیوند هیدروژنی با اسید آمینه Glu122 در مقایسه با Tyr85 برای اتصال بهتر دارای اهمیت بالاتری میباشد. Glu122 با این دو ترکیب و همچنین کلروکین پیوند هیدروژنی تشکیل داد در حالیکه با ترکیب 1 در تشکیل اتصال هیدروفوب نقش ایفا کرد. بنابراین به نظر میرسد این اسیدآمینه برای برقراری اتصال با آنزیم حائز اهمیت باشد. در مطالعات پیشین این اسیدآمینه در تشکیل پیوند هیدروژنی شرکت داشته است (15،14). ترکیب CID_11912184 یک برهمکنش هیدروژنی با اسیدآمینه Tyr85 نشان میدهد اما در کلروکین و ترکیب 1 این اسیدآمینه در تشکیل بر همکنشهای هیدروفوبی نقش دارد. در مطالعات پیشین این اسیدآمینه برای تشکیل پیوند هیدروژنی سایر ترکیبات در جایگاه فعال آنزیم pfLDH ذکر شده است (16،15). ترکیب 1 با اسیدآمینه Phe52 برهمکنش π-π تشکیل داده است در حالیکه ترکیبات CID_11912184، CID_11912187 و کلروکین با این اسیدآمینه در برهمکنشهای هیدروفوبی شرکت داشته اند؛ از اینرو احتمالاً این اسیدآمینه نقش مهمی برای اتصال در جایگاه فعال آنزیم داشته باشد. ترکیب 1 با اسیدآمینه Lys118 و Thr101 در برقراری بر همکنشهای هیدروژنی و π-کاتیون نقش دارد در حالیکه با دو ترکیب CID_11912184 و CID_11912187 و کلروکین برهمکنشی مشاهده نشد. اسیدآمینه Gly99 با ترکیب CID_11912187، کلروکین و ترکیب 1 در تشکیل بر همکنش هیدروفوب شرکت داشته است در حالیکه اسید آمینه مذکور با ترکیب CID_11912184 هیچ برهمکنشی برقرار نکرده است. در مطالعات پیشین اسیدآمینه Gly99 در تشکیل پیوند هیدروژنی ترکیبات با آنزیم شرکت داشته است (17،14). این دو ترکیب و کلروکین و ترکیب 1 با اسید آمینههای Ile54، Ile119، Phe52 و Ala98 بر همکنشهای هیدروفوبی تشکیل میدهند. ساختار دو ترکیب CID_23603310 و CID_23603337 مشابه یکدیگر است. با این تفاوت که گروه کلر در ترکیب CID_23603310 در موقعیت متا قرار گرفته است اما در ترکیب CID_23603337 در موقعیت پارا جای گرفته است. انرژی آزاد اتصال ترکیب CID_23603310 با اختلاف 23/1 کیلوکالری بر مول منفیتر از CID_23603337 است و منفیترین انرژی به دست آمده از میان 13 ترکیب حاصل از غربالگری مجازی میباشد. قرارگیری گروه کلر در موقعیت متا احتمالاً باعث تغییر کانفورماسیون ترکیب میشود که این تغییر کانفورماسیون برای اتصال با اسید آمینهها مناسب تر است. هر دو ترکیب با اسید آمینه Gly99 پیوند هیدروژنی تشکیل میدهند اما در ترکیب CID_23603310 این اسید آمینه همزمان با اتم نیتروژن حلقه پیریمیدینی و آمین گروه آمیدی پیوند هیدروژنی برقرار میکند در حالیکه با ترکیب CID_23603337 تنها با اتم نیتروژن حلقه پیریمیدینی این برهمکنش مشاهده میشود و از نظر فضایی آمین گروه آمیدی با فاصله دورتری قرار گرفته است که توانایی تشکیل این پیوند را با اسید آمینه Gly99 ندارد. بنابراین به نظر میرسد تعداد تشکیل پیوند هیدروژنی با اسیدآمینه Gly99 برای فعالیت حائز اهمیت باشد. در مطالعات پیشین این اسیدآمینه در تشکیل برهمکنش سایر ترکیبات با آنزیم مورد مطالعه شرکت داشته است (17،14). در ترکیب 1 اسیدآمینه Phe52 در تشکیل برهمکنش π-π نقش داشته است در حالیکه در دو ترکیب CID_23603310 و CID_23603337 برهمکنش هیدروفوبی تشکیل داده است. در ترکیب 1 اسیدآمینه Lys118 در ایجاد برهمکنشهای هیدروژنی و π-کاتیون شرکت داشته است در حالیکه در جایگاه اتصال آنزیم اسیدآمینه مذکور با CID_23603310 و CID_23603337 برهمکنشی نشان نداده است. ترکیب 1 با اسیدآمینه Thr101 بر همکنش هیدروژنی تشکیل میدهد در صورتیکه در ترکیب CID_23603310 در برقراری بر همکنش هیدروفوبی نقش دارد. در جایگاه فعال آنزیم با هر دو ترکیب، اسیدآمینههای Phe52، Ile119، Ile31، Thr97، Ala98 و Phe100 در تشکیل بر همکنشهای هیدروفوبی نقش دارند. در پاکت هیدروفوب، هر چهار ترکیب با اسیدآمینههای Ile119، Phe52 و Ala98 برهمکنش نشان دادند بنابراین به نظر میرسد این اسیدآمینهها در تشکیل برهمکنش هیدروفوبی مهم باشند. در مطالعات پیشین برای فعالیت بهتر ترکیبات نیز اسیدآمینههای Phe52، Val26، Ile54، Ile119، Phe100، Asn83 و Ala98 پیشنهاد شده است که نتایج حاضر را تایید میکند (20-18). برهمکنش غالب در تمام ترکیبات هیدروفوبی میباشد. بیشترین اسیدآمینهای که در تشکیل پیوند هیدروژنی نقش داردGly99 است. CID _86764490، CID_24464444، CID_11524240 و CID _14348655 با این اسیدآمینه پیوند هیدروژنی تشکیل دادهاند اما در ترکیبات CID_24342862،CID_14888646، ID_14348666،CID _14348662 ، ترکیب 1 و کلروکین این اسیدآمینه در تشکیل بر همکنش هیدروفوب نقش دارد. در مطالعات پیشین این اسیدآمینه در تشکیل پیوند هیدروژنی سایر ترکیبات با آنزیم مورد مطالعه شرکت داشته است (17،14). CID_14888646 و CID_14348662 با اسیدآمینه Glu122 که در بر همکنش کلروکین نقش داشت، پیوند هیدروژنی تشکیل دادهاند در حالیکه تمامی هفت ترکیب باقیمانده (CID_86764490، CID_24464444، CID_24342862، CID_14348666، CID_11524240، CID_989632 و CID_14348655) همانند ترکیب 1 با این اسیدآمینه برهمکنش هیدروفوبی نشان میدهند. در مطالعات پیشین این اسیدآمینه در تشکیل پیوند هیدروژنی شرکت داشته است (15،14). به نظر میرسد اسیدآمینه Glu122 در اتصال بهتر ترکیبات به جایگاه فعال آنزیم نقش مهمی داشته باشد. CID_14888646،CID _14348666 و CID_14348662 با اسیدآمینه Tyr85 پیوند هیدروژنی تشکیل دادهاند در حالیکه این اسیدآمینه با بر همکنش هیدروفوبی به ترکیبات CID_11524240، CID_989632 و CID_14348655 اتصال دارد. ترکیب CID_14888646 نیز با Tyr85 بر همکنش π-π تشکیل داده است. در مطالعات پیشین این اسیدآمینه برای تشکیل پیوند هیدروژنی نام برده شده است (16،15). ترکیب CID_86764490 با اسیدآمینه Gly29 برهمکنش هیدروژنی تشکیل میدهد در حالیکه در ترکیبات CID_24342862، CID_14348666، CID_14348662، CID_11524240 و CID_14348655 این اسیدآمینه در تشکیل بر همکنش های هیدروفوبی شرکت دارد. در ترکیبات CID_86764490، CID_24464444، CID_989632 و CID_14348655 اسیدآمینه Asp53 در تشکیل پاکت هیدروفوبی نقش دارد اما در ترکیبات CID_24342862، CID_14348666 و CID_11524240 پیوند هیدروژنی بر قرار کرده است. در مطالعات پیشین اسید آمینه مذکور در تشکیل پیوند هیدروژنی نقش ایفا کرده است (15). اسیدآمینه Thr101 در ترکیب CID_14888646 پیوند هیدروژنی برقرار کرده است در حالیکه در ترکیب CID_14348655 در ایجاد پاکت هیدروفوبی نقش داشته است. همچنین اسید آمینه Ile31 در تشکیل بر همکنشهای هیدروفوبی در جایگاه اتصال ترکیبات CID_86764490، CID_24342862 و CID_14348662 نقش دارد در حالیکه در ترکیبات CID_14348666 و CID_14348655 بر همکنش هیدروژنی برقرار کرده است. اسیدآمینه Ile54 تنها با ترکیب CID_14348666 پیوند هیدروژنی برقرار میکند در حالیکه در ترکیبات CID_24464444، CID_24342862، CID_14888646، CID_14348662، CID_989632 و CID_14348655 در ایجاد برهمکنش هیدروفوبی نقش دارد. در مطالعات پیشین برای فعالیت بهتر ترکیبات، اسیدآمینههای Phe52، Val26، Ile54، Ile119، Phe100، Asn83، Ala98 پیشنهاد شده است که همسو با نتایج حاضر میباشد (14،21). علاوه بر برهمکنشهای هیدروفوبی، پیوندهای هیدروژنی نیز در محل اتصال ترکیبات به جایگاه فعال آنزیم قابل مشاهده است که اهمیت این نوع برهمکنش را بعد از برهمکنشهای هیدروفوب نشان میدهد. با اینکه در جایگاه اتصال ترکیب 1 برهمکنش های π-کاتیون و π-π مشاهده گردید و احتمال اهمیت این نوع برهمکنش در نحوه اتصال وجود داشت، به جز ترکیب CID_14888646 هیچ یک از 13 ترکیب در تشکیل این نوع برهمکنشها نقشی نداشتند. از اینرو احتمال بر این است که وجود این نوع برهمکنشها در اتصال به آنزیم حائز اهمیت نباشد.
نتیجهگیری
بهطور خلاصه، SBVS به عنوان یک روش میتواند بهطور موفق آمیزی برای شناسایی مهارکنندههای آنزیم لاکتات دهیدروژناز پلاسمودیوم فالسیپاروم بهکار رود. در این پروژه کتابخانهای از ترکیبات با شباهت ساختاری به ترکیب 1 از پایگاه داده PubChem تشکیل گردید. ترکیبات انتخاب شده براساس معیارهای انرژی اتصال، خواص دارو همانندی و پارامترهای ADME آنالیز و غربال شدند. خواص فیزیکوشیمیایی خوب و اتصال بالا به آنزیم برای ترکیبات توصیف گردید. سپس داکینگ مولکولی برای آنالیز کیفی و کمی برهمکنشهای ترکیبات با اسید آمینههای جایگاه فعال انجام شد. از میان همه ترکیبات، CID_23603310، CID_23603337، CID_11912187 و CID_11912184 به عنوان ترکیبات الگو انتخاب و معرفی شدند. میتوان اینطور استنباط کرد که وجود حلقههای آروماتیک و بخشهای هیدروفوب،گروه آمین، پیوندهای قابل چرخش و تشکیل پیوند هیدروژنی بهینه از عوامل مهم در مهار آنزیم لاکتات دهیدروژناز پلاسمودیوم فالسیپاروم است.
سپاسگزاری
این مقاله منتج از پایاننامه داروسازی دکتری عمومی دانشگاه علوم پزشکی اردبیل است.
حامی مالی: دانشگاه علوم پزشکی اردبیل
تعارض در منافع: وجودندارد.
مالاریا کشندهترین بیماری عفونی است که بیش از 300 سال قبل آغاز گردیده اما طبق گزارش سازمان بهداشت جهانی طی دو دهه اخیر، کاهش چشمگیر موارد مرگ و میر از این بیماری مشاهده شده است. در سال 2019، 229 میلیون نفر در 87 کشور به مالاریا مبتلا و تعداد کل 409000 مرگ و میر در سراسر جهان گزارش شد (1). مالاریا توسط انگلهای تک یاختهای جنس پلاسمودیوم ایجاد و از طریق پشه آنوفل منتشر میگردد. پنج گونه شناخته شده مالاریا شامل پلاسمودیوم مالاریا، پلاسمودیوم اوالا، پلاسمودیوم ویواکس، پلاسمودیوم ناولسکی و پلاسمودیوم فالسیپاروم وجود دارند. از بین پنج گونه انگلی پلاسمودیوم، پلاسمودیوم فالسیپاروم انسان را آلوده میکند و از میان همه آنها کشندهتر است (2). در غیاب یک واکسن موثر، استفاده از روشهای شیمیدرمانی به عنوان عوامل ضد مالاریا تنها راه باقیمانده برای مدیریت و پیشگیری از بیماری مالاریا است. تحقیقات نشان دادند که اثربخشی بیشتر ترکیبات ضد مالاریا به گونههای پلاسمودیوم مقاومت نشان میدهند. مقاومت در برابر تمام گروههای اصلی داروها شامل 4-آمینوکوئینولینها (کلروکین، آمودیاکین و پیپراکین)، آنتی فولاتها، آریل آمینو الکلها (کینین، لومفانترین و مفلوکین)، مشتقات آرتمیزینین، آنتیبیوتیکها (کلیندامایسین و داکسی سایکلین) و نپتوکوکینون گزارش شده است (3). این امر باعث میشود محققان بر روی اهداف پلاسمودیوم فالسیپاروم (pf) کار کنند تا داروهای موثر بر ضد آن و سایر گونههای مالاریا را که بر انسان موثر است معرفی نمایند. کلروکین بهطور وسیعی از دهه 1940 به عنوان داروی انتخابی با هزینه قابل قبول برای درمان همه انواع مالاریا استفاده شده است. ویژگیهای مناسب کلروکین باعث استفاده گسترده از آن و نهایتاً مقاومت به آن در pf منجر شد. در نتیجه این گونه انگل مسئول گسترده ترین مشکلات مالاریای انسانی گردید (4). این امر نیاز به توسعه داروهای ضد مالاریا جدید را افزایش داده است که در کنترل مالاریا موثرتر باشد. اهداف مختلفی برای داروهای ضد مالاریا مانند پلاسمپسین I، II و V، فالسیپایین2، لاکتات دهیدروژناز pf و پیریدوکسال کیناز وجود دارد. از میان این اهداف، آنزیم لاکتات دهیدروژناز (LDH) یک هدف ارجح برای داروهای ضد مالاریا است؛ زیرا تولید آدنوزین تری فسفات (ATP) در پلاسمودیوم را از طریق مسیر گلیکولیتیک کنترل میکند. مسیرهای گلیکولیتیک و آنزیمهای مرتبط به دلیل وابستگی انگلی آنها به چرخه گلیکولیز برای تولید انرژی، اهداف دارویی حیاتی هستند (5). pfLDH یک نقش مهم در آخرین مرحله گلیکولیز دارد و تبدیل پیروات به لاکتات را کاتالیز میکند. آنزیم pfLDH بیشتر با توسعه NAD+ مرتبط است که برای آنزیم گلیکولیتیک گلیسرآلدئید-3-فسفات دهیدروژناز مورد نیاز است (6). انگلهای pf برای تولید انرژی خود به این آنزیم نیازمند هستند که برای عملکرد، رشد و توسعه بیوشیمیایی مهم است. بنابراین، این آنزیم یک هدف دارویی مهم در درمان مالاریا است و مهار آن باعث مرگ انگل میگردد. توالی اسیدهای آمینه برای LDH انسانی و pfLDH شباهت کمی وجود دارد (7). بنابراین، هدفگیری انتخابی این آنزیم گلیکولیتیک در pfLDH قابلتوجه است اما LDH انسانی را متوقف نمیکند. این ویژگیها، pfLDH را به عنوان یک هدف مناسب برای طراحی مبتنی بر ساختار ضد مالاریاهای جدید توصیه میکند. غربالگری مجازی (VS) یک استراتژی کاربردی است که برای تمایز مولکولها براساس ویژگی مورد نظر استفاده میشود و میتواند برای شناسایی ترکیبات الگوی جدید مفید باشد. میتوان آن را به دو دسته گسترده تقسیم کرد: مبتنی بر ساختار و مبتنی بر لیگاند (8). روشهای غربالگری مجازی مبتنی بر ساختار یا گیرنده (SBVS) زمانی بسیار مناسب هستند که اطلاعات مربوط به ساختار هدف در دسترس باشد. در اینروش حالتهای اتصال برای هر لیگاند پیشبینی میشود. رویکردهای غربالگری مجازی مبتنی بر لیگاند (LBVS) از اطلاعات دادههای ساختار-فعالیت مولکولهای فعال شناساییشده با هدف شناسایی ترکیبات ساختاری متنوع با زیست فعالیتی مشابه استفاده میکنند. ترکیبات الگوی شناسایی شده از VS، به عنوان نقطه شروعی برای بهینهسازی ترکیبات الگو در برنامههای کشف دارو میباشد (9). در این مطالعه، به منظور شناسایی و معرفی مهارکنندههای بالقوه جهت مهار آنزیم pfLDH با استفاده از روشهای غربالگری مجازی مبتنی بر ساختار، مهارکنندههای احتمالی آنزیم pfLDH به عنوان ترکیبات دارویی ضد مالاریا طراحی شدند. بدین منظور کتابخانهای از ترکیبات تشکیل شد و با بهرهگیری از فیلتراسیونهای مختلف ترکیبات مورد ارزیابی قرار گرفتند. فلوچارت فرایند VS در شکل 1 نشان داده شده است.
شکل 1: فلوچارت فرایند کلی غربالگری مجازی
روش بررسی
این پژوهش به صورت توصیفی-تحلیلی صورت گرفت. در این مطالعه ترکیب 7-((4-(6،2-دی اکسو-7،3،1-تری متیل-7،6،3،2- تتراهیدرو-H1-پورین-8-یل)اکسی)-3-متوکسی فنیل)-3-متیل-1-فنیل-5-(5،4،3-تری متوکسی فنیل)-8،7،6،1-تتراهیدروپیرازولو [4،3-b][4،1] دیازپین (ترکیب 1، شکل 2) با فعالیت ضد مالاریای قوی (مقدار IC50 برابر 3/11 ماکروگرم بر میلیلیتر و درصد مهار برابر 94/2%) که در مطالعات قبلی گزارش شده بود (10) به عنوان ترکیب الگو برای تهیه کتابخانه ترکیبات مورد استفاده قرار گرفت.
شکل 2. ساختار شیمیایی ترکیب 1
تهیه کتابخانه داده و غربالگری مجازی: قبل از شروع غربالگری مجازی، ابتدا لازم است کتابخانهای از ترکیبات تهیه شود. در این پژوهش ابتدا کتابخانه ترکیبات از پایگاه داده PubChem استخراج گردید (https://pubchem.ncbi.nlm.nih.gov). پایگاه PubChem یک پایگاه رایگان با بیش از 100 میلیون ساختار است. کتابخانه شامل 8733 ترکیب از پایگاه داده PubChem تشکیل شد. همه ساختارها در فورمت SDF ذخیره و سپس به فورمت pdbqt توسط نرمافزار PyRx 0/8 (https://pyrx.sourceforge.io) تبدیل شدند. در مرحله بعد، ترکیبات کتابخانه داخل جایگاه فعال آنزیم pfLDH با استفاده از اتوداک وینا در نرمافزار PyRx 0/8 داک گردیدند. تعداد runها برای هر آنالیز داکینگ روی 100 تنظیم شد. الگوریتم بهکار برده شده لامارکین ژنتیک بود. نتایج داکینگ براساس انرژی آزاد اتصال مرتب و ترکیبات با بالاترین انرژی آزاد اتصال انتخاب گردیدند.
بررسی خواص داروهمانندی: از میان کل ترکیبات کتابخانه، 1001 ترکیب از مرحله قبل با بالاترین انرژی آزاد اتصال انتخاب شدند و برای ارزیابی خواص داروهمانندی و پارامترهای قانون پنج لیپنیسکی وارد این مرحله گردیدند. پارامترهای مورد ارزیابی شامل وزن مولکولی، تعداد اتمهای دهنده هیدروژن، تعداد اتمهای گیرنده هیدروژن، لیپوفیلیسیته (LogP)، توپولوژی مساحت سطح قطبی (TPSA) و تعداد پیوندهای قابل چرخش است. تمام این خواص با استفاده از وب سرور Molinspiration محاسبه شد (https://www.molinspiration.com).
بررسی ویژگیهای فارماکوکینتیکی ترکیبات (ADMET): خواص فارماکوکینتیک شناخته شده تحت عنوان ADMET (جذب، پخش، متابولیسم، دفع و سمیت) موقعیت ترکیبات دارویی را در ارگانیسم بررسی میکنند. یک داروی موفق تنها منوط به داشتن قدرت اثر خوب نیست بلکه باید ویژگیهای ADMET قابل قبولی داشته باشد. بنابراین در این مرحله خواص ADMET 281 ترکیب راه یافته از مرحله قبل توسط وب سرورهای swiss ADME (http://www.swissadme.ch) و admet SAR (http://lmmd.ecust.edu.cn/admetsar2) مورد بررسی قرار گرفت. در نهایت از 281 ترکیب وارد شده 13 ترکیب توانستند پروفایل فارماکوکینتیک خوبی را نشان دهند و به مرحله بعدی راه پیدا کنند.
مطالعه شبیه سازی داکینگ مولکولی: برای انجام روش داکینگ مولکولی از نرمافزار اتوداک 4.2 استفاده شد (11). بدین منظور نرمافزار اتوداک بر روی کامپیوتر 8 هستهای تحت سیستم عامل ویندوز نصب گردید. ژنتیک الگوریتم (GA) به عنوان الگوریتم جستجوگر توسط نرمافزار مورد استفاده قرار گرفت. برنامه گرافیکی اتوداک تولز 6.5.1 (ADT) برای تهیه، انجام و آنالیز شبیهسازیهای داکینگ مولکولی بهکار رفت. در ابتدا، ساختارهای دوبعدی ترکیبات بهوسیله برنامه ChemDraw Ultra 10.0 ترسیم (12) و سپس با استفاده از نرمافزار Hyperchem8 HyperChem, Release 8.0 for Windows, Molecular Modeling System: HyperCube, 2007)) در میدان نیروی مکانیک مولکولی (MM+) و روش نیمه تجربی PM3 و الگوریتم Polak-Ribiere از نظر انرژی بهینه شدند. پس از بهینهسازی انرژی لیگاند، با استفاده از نرمافزار اتوداک تولز اتمهای هیدروژن به ساختار مولکول افزوده شدند. در مرحله بعد، اتمهای هیدروژن غیرقطبی در اتم کربن مربوطه ادغام و بار الکتریکی گستیگر (بارهای الکتریکی اتم که بهصورت تجربی محاسبه میگردد) و تعداد درجات آزادی زوایای پیچشی لیگاند با استفاده از نرمافزار اتوداک تولز محاسبه گردید. در نهایت فایل لیگاند به صورت pdbqt ذخیره شد. ساختار کریستالی سه بعدی آنزیم pfLDH از پایگاه بانک دادههای پروتئین دانلود گردید (کد شناسایی 1CET) (https://www.rcsb.org). روش انجام داکینگ و آنالیز کانفورمرها براساس توضیحات در مقالات قبلی صورت گرفت (13). در ابتدا با استفاده از نرمافزار Notpat++ و یا Discovery Studio Viewer lite 4.0 مولکولهای آب از ساختار کریستالوگرافی حذف گردیدند. سپس با استفاده از نرمافزار اتوداک تولز اتمهای هیدروژن به ساختار کریستالوگرافی افزوده شدند. در مرحله بعد، اتمهای هیدروژن غیر قطبی در اتم کربن مربوطه ادغام شده و بار الکتریکی کلمن و پارامترهای حلال پوشی ماکرومولکول محاسبه و در نهایت به صورت pdbqt ذخیره گردید. پس از تهیه فایلهای ورودی مورد نیاز داکینگ (ماکرومولکول، لیگاند و نقشه اتصال)، مطالعات داکینگ به منظور مدل سازی برهمکنشهای لیگاند-گیرنده، با استفاده از الگوریتمی تحت عنوان ژنتیک لامارکین انجام شد. در مرحله بعد، براساس حجم مولکولی لیگاندهای طراحی شده، شبکهای با ابعاد 60 × 60 × 60 آنگستروم در راستای محورهای سه گانه مختصات و فاصله نقاط شبکه 0/573 آنگستروم (یک چهارم طول پیوند ساده کربن-کربن) که در برگیرنده جایگاه فعالگیرنده بود، در نظر گرفته شد. فایل شبکه به صورت gpf ذخیره گردید. پارامترهای ذخیره شده در فایل gpf در اختیار محاسبات اتوگرید قرار گرفته است. پس از انجام عملیات داکینگ، نتایج شامل کانفورماسیونهای مولکول، انواع برهمکنشهای مولکول با پروتئین شامل برهمکنشهای هیدروژنی، هیدروفوبی و π-π با اسیدهای آمینه موجود در جایگاه اتصال پروتئینها قابل مشاهده و تجزیه و تحلیل میباشند. به منظور دستیابی به این اطلاعات از نرمافزارهای اتوداک تولز و Discovery Studio Viewer lite 4.0 استفاده شد (Accelryslnc, San Diego, CA, USA).
تجزیه و تحلیل آماری
در این مطالعه ترکیبات با پتاسیل مهار آنزیم pfLDH براساس روش غربالگری مجازی مبتنی بر ساختار شناسایی و معرفی گردید.
ملاحظات اخلاقی
کد اخلاق مصوبه این پروژه توسط دانشگاه علوم پزشکی اردبیل IR.ARUMS.REC.1397.138 است.
نتایج
تشکیل کتابخانه و غربالگری مجازی: در زمینه کشف و طراحی ترکیبات دارویی، VS میتواند به طور موثری برای انتخاب مولکولهای زیست فعال بالقوه از کتابخانه ترکیبات برای اتصال به پروتئین هدف استفاده شود. در این پروژه از پایگاه داده PubChem برای جستجوی ترکیبات با شباهت 70% به ترکیب 1 استفاده شد. نتیجه این جستجو براساس شباهت ساختاری، منجر به تشکیل یک کتابخانه با 8733 ترکیب گردید. سپس VS بر روی ترکیبات جمعآوری شده توسط نرمافزار PyRx انجام شد. بعد از اتمام کار 1001 ترکیب با انرژی آزاد اتصال برابر و بالاتر از Kcal/mol 8/79- برای مرحله بعد انتخاب شدند (برابر و بالاتر از انرژی آزاد اتصال ترکیب 1، Kcal/mol 79/8-).
خواص داروهمانندی: یکی از فاکتورهای مهم برای کشف و پیشرفت ترکیبات زیست فعال به عنوان یک داروی خوراکی فراهمی زیست بالای آنها است. برای پیش بینی ترکیبات زیست فعال به عنوان یک داروی خوراکی خوب باید معیارهای زیر را مورد توجه قرار داد: پیوندهای قابل چرخش مولکول که تحت عنوان انعطافپذیری مولکول شناخته میشود، جذب گوارشی خوب و TPSA پایین (مجموع پیوندهای هیدروژنیدهنده و پذیرنده). علاوه بر آنها، قانون پنج لیپنسکی ویژگیهای داروهمانندی شامل جرم مولکولی، مقادیر پیوندهای هیدروژنی دهنده و پذیرنده و LogP را معرفی میکند. مجموع این معیارها کمک میکند که ترکیباتی با خصوصیات فارماکوکینتیک بهتر در بدن انسان برای تجویز خوراکی معرفی گردد. در این فاز، پارامترهای فیزیکوشیمیایی مانند CLogP، TPSA، جرم مولکولی، پیوندهای هیدروژنی دهنده و گیرنده، و پیوندهای قابل چرخش بر روی ترکیبات انتخاب شده از مرحله قبل پیادهسازی شد. نهایتاً، 281 ترکیب از معیارهای ذکر شده تبعیت کردند و خواص داروهمانندی مناسبی را نشان دادند.
خواص فارماکوکینتیک (ADMET): محاسبه خواص فارماکوکینتیک ترکیبات در شناسایی اولیه آنها به عنوان ترکیبات الگو قابلتوجه میباشد. بررسی این خواص منجر به حذف کاندیدهای دارویی ضعیف میشود. در این مرحله خواص فیزیکوشیمیایی و فارماکوکینتیکی ترکیبات انتخاب شده توسط وب سرورهای swissADME و admetSAR بررسی گردید. پارامترهای مورد بررسی شامل مقدار لیپوفیلیسیتی، نفوذپذیری سد خونی-مغزی (BBB)، حلالیت، جذب و متابولیسم و همچنین مهار آنزیمهای کبدی و p-گلیکوپروتئین میباشد. در این مرحله، توسط وب سرورهای swissADME و admetSAR خواص ADMET 281 ترکیب منتخب از مرحله قبل مورد بررسی قرار گرفت. از این تعداد، 13 ترکیب با بهترین خواص فارماکوکینتیک انتخاب شدند. نتایج خواص دارو همانندی و فارماکوکینتیک 13 ترکیب منتخب در جداول 1 و 2 نشان داده شده است.
جدول 1: خواص داروهمانندی ترکیبات منتخب حاصل از غربالگری مجازی
TPSA: سطح توپولوژیک قطبی؛ MW: جرم مولکولی؛ NON: تعداد پیوندهای هیدروژنی پذیرنده؛ NOHNH: تعداد پیوندهای هیدروژنی دهنده؛ nROTB: تعداد پیوندهای قابل چرخش
جدول 2. ویژگیهای فارماکوکینتیکی ترکیبات منتخب حاصل از غربالگری مجازی
GI: جذب گوارشی؛ BBB: سد خونی-مغزی؛ P-gp: P-گلیکوپروتئین
مطالعات شبیهسازی داکینگ مولکولی: شبیهسازی داکینگ مولکولی برای مطالعه حالتهای اتصال ترکیبات منتخب داخل جایگاه فعال آنزیم انجام شد. در ابتدا به منظور اعتبار سنجی پروتکل داکینگ، لیگاند کلروکین (کوکریستال) از جایگاه فعال آنزیم حذف شد و مجدداً در جایگاه فعال داک گردید. تجزیه و تحلیل نتایج تحت عنوان ریشه انحراف میانگین مربع (RMSD) بین حالتهای داک شده و کریستالوگرافی شده انجام گردید. مقدار RMSD بهدست آمده بین کانفورماسیون داک شده کلروکین و حالت کریستال شده برابر 1/03 آنگستروم بود. براساس شاخص بینالمللی مقادیر زیر 2 آنگستروم مورد پذیرش میباشد. بعد از اعتبارسنجی پروتکل داکینگ، بررسی نتایج داکینگ 13 ترکیب منتخب از مرحله قبل نشان داد که تمام این ترکیبات فضای مشابه را درون جایگاه فعال آنزیم اشغال میکنند. ساختار شیمیایی این 13 ترکیب در شکل 3 نشان داده شده است.
داکینگ مولکولی 13 ترکیب حاصل از غربالگری مجازی در جایگاه فعال آنزیم pfLDH با کد شناسایی 1CET در نرمافزار داکینگ انجام گردید (جدول 3).
بالاترین انرژی آزاد اتصال ( Kcal/mol29/10-) برای برهمکنش CID_23603310 با اسید آمینههای جایگاه فعال به صورت زیر تفسیر میشود (شکل 4): دو پیوند هیدروژنی بین اتم نیتروژن حلقه پیریمیدین و NH آمیدی ترکیب و اسید آمینه Gly99 تشکیل شد. برهمکنشهای هیدروفوب لیگاند با اسید آمینههای Met30، Ile31، Gly27، Ile119، Asp53، Phe52، Thr101، Phe100، Ala98، Thr97 و Gly99 مشاهده گردید. ترکیب CID_23603337 با انرژی آزاد اتصال 9/06- کیلوکالری بر مول برهمکنشهای زیر را نشان داد (شکل 5): یک پیوند هیدروژنی بین اتم نیتروژن حلقه پیریمیدین و اسید آمینه Gly99 تشکیل شد. اسیدآمینههای Phe100، Thr97، Ala98، Val138، Val26، Phe52، Ile119، Tyr85 و Ile31 با ترکیب CID_23603337 برهمکنشهای هیدروفوب تشکیل دادند. حالت اتصال CID_11912187 در جایگاه فعال آنزیم در شکل 6 مشاهده میشود. این ترکیب دو پیوند هیدروژنی نشان داد؛ دو پیوند هیدروژنی بین NH گروه آمیدی متصل به حلقه فنیل و NH متصل به حلقه پیریمیدین و اسید آمینه Glu122. این ترکیب همچنین برهمکنشهای هیدروفوب با اسید آمینه های Ile121، Ile119، Phe52، Ile54، Ala98، Gly99 و Phe100 تشکیل داد. ترکیب CID_11912184 با انرژی آزاد اتصال 9/00- کیلوکالری بر مول دو پیوند هیدروژنی تشکیل دادند (شکل 7): یک پیوند هیدروژنی بین اتم اکسیژن بخش دی اکسولون با اسیدآمینه Tyr85 و یک پیوند هیدروژنی بین NH متصل به حلقه پیریمیدین با اسید آمینه Glu122. اسید آمینههای Ile54، His126، Ile119، Val26، Phe52، Gly27، Ala98 و Asp53 با ترکیب برهمکنش هیدروفوب تشکیل دادند.
شکل 3: ساختارهای شیمیایی 13 ترکیب منتخب راه یافته به مرحله داکینگ مولکولی
جدول 3: انرژی آزاد اتصال و برهمکنشهای هیدروژنی، هیدروفوبی و π-π ترکیبات منتخب حاصل از غربالگری مجازی در جایگاه فعال آنزیم pfLDH
شکل 4: حالت اتصال و برهمکنشهای (A) سه بعدی (B) دو بعدی ترکیب CID_23603310 در جایگاه فعال آنزیم pfLDH
شکل 5: حالت اتصال و برهمکنشهای (A) سه بعدی (B) دو بعدی ترکیب CID_23603337 در جایگاه فعال آنزیم pfLDH
شکل 6: حالت اتصال و برهمکنشهای (A) سه بعدی (B) دو بعدی ترکیب CID_11912187 در جایگاه فعال آنزیم pfLDH
شکل 7: حالت اتصال و برهمکنشهای (A) سه بعدی (B) دو بعدی ترکیب CID_11912184 در جایگاه فعال آنزیم pfLDH
بحث
دو معیار مهم در تعیین بهترین حالت داک شده، بیشترین (منفیترین) انرژی آزاد اتصال تخمین زده شده و همچنین بیشترین برهمکنشهای مناسب با اسید آمینههای اصلی جایگاه فعال آنزیم pfLDH میباشند. 13 ترکیب نهایی با خصوصیات داروهمانندی و فارماکوکینتیک مناسب به مرحله شبیهسازی داکینگ مولکولی راه یافتند. طبق نتایج حاصل از داکینگ مولکولی همه چهار ترکیب مذکور انرژی آزاد اتصال منفیتری نسبت به کلروکین نشان داده اند. ساختار دو ترکیب CID_11912184 و CID_11912187 مشابه یکدیگر است. ساختار CID_11912187 یک گروه متیل اضافه نسبت به CID_11912184 دارد. این گروه متیل لیپوفیل به نظر میآید تا حدی اثر مثبت روی اتصال ترکیب در جایگاه فعال آنزیم داشته باشد. هر دو این ترکیبات انرژی آزاد اتصال بهتری نسبت به کلروکین و ترکیب 1 نشان دادند. ترکیب CID_11912184 یک پیوند هیدروژنی و ترکیب CID_1912187 دو پیوند هیدروژنی با اسیدآمینه Glu122 تشکیل میدهند. در ترکیب CID_11912187 گروه NH متصل به حلقه پیریمیدینی و آمیدی هر دو با Glu122 پیوند هیدروژنی تشکیل میدهند در حالیکه ترکیب CID_11912184 تنها NH متصل به حلقه پیریمیدینی با این اسید آمینه پیوند هیدروژنی تشکیل میدهد. علاوه بر آن این ترکیب از سر اکسیژن بنزودی اکسول با اسید آمینه Tyr85 پیوند هیدروژنی تشکیل میدهد. با توجه به نتایج مشاهده شده و مطالعات قبلی تشکیل پیوند هیدروژنی با اسید آمینه Glu122 در مقایسه با Tyr85 برای اتصال بهتر دارای اهمیت بالاتری میباشد. Glu122 با این دو ترکیب و همچنین کلروکین پیوند هیدروژنی تشکیل داد در حالیکه با ترکیب 1 در تشکیل اتصال هیدروفوب نقش ایفا کرد. بنابراین به نظر میرسد این اسیدآمینه برای برقراری اتصال با آنزیم حائز اهمیت باشد. در مطالعات پیشین این اسیدآمینه در تشکیل پیوند هیدروژنی شرکت داشته است (15،14). ترکیب CID_11912184 یک برهمکنش هیدروژنی با اسیدآمینه Tyr85 نشان میدهد اما در کلروکین و ترکیب 1 این اسیدآمینه در تشکیل بر همکنشهای هیدروفوبی نقش دارد. در مطالعات پیشین این اسیدآمینه برای تشکیل پیوند هیدروژنی سایر ترکیبات در جایگاه فعال آنزیم pfLDH ذکر شده است (16،15). ترکیب 1 با اسیدآمینه Phe52 برهمکنش π-π تشکیل داده است در حالیکه ترکیبات CID_11912184، CID_11912187 و کلروکین با این اسیدآمینه در برهمکنشهای هیدروفوبی شرکت داشته اند؛ از اینرو احتمالاً این اسیدآمینه نقش مهمی برای اتصال در جایگاه فعال آنزیم داشته باشد. ترکیب 1 با اسیدآمینه Lys118 و Thr101 در برقراری بر همکنشهای هیدروژنی و π-کاتیون نقش دارد در حالیکه با دو ترکیب CID_11912184 و CID_11912187 و کلروکین برهمکنشی مشاهده نشد. اسیدآمینه Gly99 با ترکیب CID_11912187، کلروکین و ترکیب 1 در تشکیل بر همکنش هیدروفوب شرکت داشته است در حالیکه اسید آمینه مذکور با ترکیب CID_11912184 هیچ برهمکنشی برقرار نکرده است. در مطالعات پیشین اسیدآمینه Gly99 در تشکیل پیوند هیدروژنی ترکیبات با آنزیم شرکت داشته است (17،14). این دو ترکیب و کلروکین و ترکیب 1 با اسید آمینههای Ile54، Ile119، Phe52 و Ala98 بر همکنشهای هیدروفوبی تشکیل میدهند. ساختار دو ترکیب CID_23603310 و CID_23603337 مشابه یکدیگر است. با این تفاوت که گروه کلر در ترکیب CID_23603310 در موقعیت متا قرار گرفته است اما در ترکیب CID_23603337 در موقعیت پارا جای گرفته است. انرژی آزاد اتصال ترکیب CID_23603310 با اختلاف 23/1 کیلوکالری بر مول منفیتر از CID_23603337 است و منفیترین انرژی به دست آمده از میان 13 ترکیب حاصل از غربالگری مجازی میباشد. قرارگیری گروه کلر در موقعیت متا احتمالاً باعث تغییر کانفورماسیون ترکیب میشود که این تغییر کانفورماسیون برای اتصال با اسید آمینهها مناسب تر است. هر دو ترکیب با اسید آمینه Gly99 پیوند هیدروژنی تشکیل میدهند اما در ترکیب CID_23603310 این اسید آمینه همزمان با اتم نیتروژن حلقه پیریمیدینی و آمین گروه آمیدی پیوند هیدروژنی برقرار میکند در حالیکه با ترکیب CID_23603337 تنها با اتم نیتروژن حلقه پیریمیدینی این برهمکنش مشاهده میشود و از نظر فضایی آمین گروه آمیدی با فاصله دورتری قرار گرفته است که توانایی تشکیل این پیوند را با اسید آمینه Gly99 ندارد. بنابراین به نظر میرسد تعداد تشکیل پیوند هیدروژنی با اسیدآمینه Gly99 برای فعالیت حائز اهمیت باشد. در مطالعات پیشین این اسیدآمینه در تشکیل برهمکنش سایر ترکیبات با آنزیم مورد مطالعه شرکت داشته است (17،14). در ترکیب 1 اسیدآمینه Phe52 در تشکیل برهمکنش π-π نقش داشته است در حالیکه در دو ترکیب CID_23603310 و CID_23603337 برهمکنش هیدروفوبی تشکیل داده است. در ترکیب 1 اسیدآمینه Lys118 در ایجاد برهمکنشهای هیدروژنی و π-کاتیون شرکت داشته است در حالیکه در جایگاه اتصال آنزیم اسیدآمینه مذکور با CID_23603310 و CID_23603337 برهمکنشی نشان نداده است. ترکیب 1 با اسیدآمینه Thr101 بر همکنش هیدروژنی تشکیل میدهد در صورتیکه در ترکیب CID_23603310 در برقراری بر همکنش هیدروفوبی نقش دارد. در جایگاه فعال آنزیم با هر دو ترکیب، اسیدآمینههای Phe52، Ile119، Ile31، Thr97، Ala98 و Phe100 در تشکیل بر همکنشهای هیدروفوبی نقش دارند. در پاکت هیدروفوب، هر چهار ترکیب با اسیدآمینههای Ile119، Phe52 و Ala98 برهمکنش نشان دادند بنابراین به نظر میرسد این اسیدآمینهها در تشکیل برهمکنش هیدروفوبی مهم باشند. در مطالعات پیشین برای فعالیت بهتر ترکیبات نیز اسیدآمینههای Phe52، Val26، Ile54، Ile119، Phe100، Asn83 و Ala98 پیشنهاد شده است که نتایج حاضر را تایید میکند (20-18). برهمکنش غالب در تمام ترکیبات هیدروفوبی میباشد. بیشترین اسیدآمینهای که در تشکیل پیوند هیدروژنی نقش داردGly99 است. CID _86764490، CID_24464444، CID_11524240 و CID _14348655 با این اسیدآمینه پیوند هیدروژنی تشکیل دادهاند اما در ترکیبات CID_24342862،CID_14888646، ID_14348666،CID _14348662 ، ترکیب 1 و کلروکین این اسیدآمینه در تشکیل بر همکنش هیدروفوب نقش دارد. در مطالعات پیشین این اسیدآمینه در تشکیل پیوند هیدروژنی سایر ترکیبات با آنزیم مورد مطالعه شرکت داشته است (17،14). CID_14888646 و CID_14348662 با اسیدآمینه Glu122 که در بر همکنش کلروکین نقش داشت، پیوند هیدروژنی تشکیل دادهاند در حالیکه تمامی هفت ترکیب باقیمانده (CID_86764490، CID_24464444، CID_24342862، CID_14348666، CID_11524240، CID_989632 و CID_14348655) همانند ترکیب 1 با این اسیدآمینه برهمکنش هیدروفوبی نشان میدهند. در مطالعات پیشین این اسیدآمینه در تشکیل پیوند هیدروژنی شرکت داشته است (15،14). به نظر میرسد اسیدآمینه Glu122 در اتصال بهتر ترکیبات به جایگاه فعال آنزیم نقش مهمی داشته باشد. CID_14888646،CID _14348666 و CID_14348662 با اسیدآمینه Tyr85 پیوند هیدروژنی تشکیل دادهاند در حالیکه این اسیدآمینه با بر همکنش هیدروفوبی به ترکیبات CID_11524240، CID_989632 و CID_14348655 اتصال دارد. ترکیب CID_14888646 نیز با Tyr85 بر همکنش π-π تشکیل داده است. در مطالعات پیشین این اسیدآمینه برای تشکیل پیوند هیدروژنی نام برده شده است (16،15). ترکیب CID_86764490 با اسیدآمینه Gly29 برهمکنش هیدروژنی تشکیل میدهد در حالیکه در ترکیبات CID_24342862، CID_14348666، CID_14348662، CID_11524240 و CID_14348655 این اسیدآمینه در تشکیل بر همکنش های هیدروفوبی شرکت دارد. در ترکیبات CID_86764490، CID_24464444، CID_989632 و CID_14348655 اسیدآمینه Asp53 در تشکیل پاکت هیدروفوبی نقش دارد اما در ترکیبات CID_24342862، CID_14348666 و CID_11524240 پیوند هیدروژنی بر قرار کرده است. در مطالعات پیشین اسید آمینه مذکور در تشکیل پیوند هیدروژنی نقش ایفا کرده است (15). اسیدآمینه Thr101 در ترکیب CID_14888646 پیوند هیدروژنی برقرار کرده است در حالیکه در ترکیب CID_14348655 در ایجاد پاکت هیدروفوبی نقش داشته است. همچنین اسید آمینه Ile31 در تشکیل بر همکنشهای هیدروفوبی در جایگاه اتصال ترکیبات CID_86764490، CID_24342862 و CID_14348662 نقش دارد در حالیکه در ترکیبات CID_14348666 و CID_14348655 بر همکنش هیدروژنی برقرار کرده است. اسیدآمینه Ile54 تنها با ترکیب CID_14348666 پیوند هیدروژنی برقرار میکند در حالیکه در ترکیبات CID_24464444، CID_24342862، CID_14888646، CID_14348662، CID_989632 و CID_14348655 در ایجاد برهمکنش هیدروفوبی نقش دارد. در مطالعات پیشین برای فعالیت بهتر ترکیبات، اسیدآمینههای Phe52، Val26، Ile54، Ile119، Phe100، Asn83، Ala98 پیشنهاد شده است که همسو با نتایج حاضر میباشد (14،21). علاوه بر برهمکنشهای هیدروفوبی، پیوندهای هیدروژنی نیز در محل اتصال ترکیبات به جایگاه فعال آنزیم قابل مشاهده است که اهمیت این نوع برهمکنش را بعد از برهمکنشهای هیدروفوب نشان میدهد. با اینکه در جایگاه اتصال ترکیب 1 برهمکنش های π-کاتیون و π-π مشاهده گردید و احتمال اهمیت این نوع برهمکنش در نحوه اتصال وجود داشت، به جز ترکیب CID_14888646 هیچ یک از 13 ترکیب در تشکیل این نوع برهمکنشها نقشی نداشتند. از اینرو احتمال بر این است که وجود این نوع برهمکنشها در اتصال به آنزیم حائز اهمیت نباشد.
نتیجهگیری
بهطور خلاصه، SBVS به عنوان یک روش میتواند بهطور موفق آمیزی برای شناسایی مهارکنندههای آنزیم لاکتات دهیدروژناز پلاسمودیوم فالسیپاروم بهکار رود. در این پروژه کتابخانهای از ترکیبات با شباهت ساختاری به ترکیب 1 از پایگاه داده PubChem تشکیل گردید. ترکیبات انتخاب شده براساس معیارهای انرژی اتصال، خواص دارو همانندی و پارامترهای ADME آنالیز و غربال شدند. خواص فیزیکوشیمیایی خوب و اتصال بالا به آنزیم برای ترکیبات توصیف گردید. سپس داکینگ مولکولی برای آنالیز کیفی و کمی برهمکنشهای ترکیبات با اسید آمینههای جایگاه فعال انجام شد. از میان همه ترکیبات، CID_23603310، CID_23603337، CID_11912187 و CID_11912184 به عنوان ترکیبات الگو انتخاب و معرفی شدند. میتوان اینطور استنباط کرد که وجود حلقههای آروماتیک و بخشهای هیدروفوب،گروه آمین، پیوندهای قابل چرخش و تشکیل پیوند هیدروژنی بهینه از عوامل مهم در مهار آنزیم لاکتات دهیدروژناز پلاسمودیوم فالسیپاروم است.
سپاسگزاری
این مقاله منتج از پایاننامه داروسازی دکتری عمومی دانشگاه علوم پزشکی اردبیل است.
حامی مالی: دانشگاه علوم پزشکی اردبیل
تعارض در منافع: وجودندارد.
References:
1- World Malaria Report. 2020. Available at: https://www.who.int/publications/i/item/9789240015791. Accessed March 2021.
2- Muhseen ZT, Hameed AR, Al-Bhadly O, Ahmad S, Li G. Natural Products for Treatment of Plasmodium Falciparum Malaria: An Integrated Computational Approach. Comput Biol Med 2021; 134: 104415.
3- Varela-Aramburu S, Ghosh C, Goerdeler F, Priegue P, Moscovitz O, Seeberger PH. Targeting and Inhibiting Plasmodium Falciparum Using Ultra-Small Gold Nanoparticles. ACS Appl Mater Interfaces 2020; 12(39): 43380-87.
4- Flegg JA, Metcalf CJE, Gharbi M, Venkatesan M, Shewchuk T, Sibley CH, Guerin PJ. Trends in Antimalarial Drug Use in Africa. Am J Trop Med 2013; 89(5): 857-65.
5- Waingeh, VF, Groves AT, Eberle JA. Binding of Quinoline-Based Inhibitors to Plasmodium falciparum Lactate Dehydrogenase: A Molecular Docking Study. Open J Biophys 2013; 3(4): 285-90.
6- Penna-Coutinho J, Cortopassi WA, Oliveira AA, França TCC, Krettli AU. Antimalarial Activity of Potential Inhibitors of Plasmodium falciparum Lactate Dehydrogenase Enzyme Selected by Docking Studies. PLoS ONE 2011; 6(7): e21237.
7- Singh R, Bhardwaj V, Purohit R. Identification of a Novel Binding Mechanism of Quinoline Based Molecules with Lactate Dehydrogenase of Plasmodium Falciparum. J Biomol Struct Dyn 2021; 39(1): 348-56.
8- Reynolds CR, Muggleton SH, Sternberg MJE. Incorporating Virtual Reactions into a Logic-based Ligand-based Virtual Screening Method to Discover New Leads. Mol Inform 2015; 34(9): 615-25.
9- Sepehri S, Saghaie L, Fassihi A. Anti-HIV-1 Activity Prediction of Novel Gp41 Inhibitors Using Structure-Based Virtual Screening and Molecular Dynamics Simulation. Mol Inform 2017; 36(3): 1600060.
10- Insuasty B, Ramírez J, Becerra D, Echeverry C, Quiroga J, Abonia R, et al. An Efficient Synthesis of New Caffeine-Based Chalcones, Pyrazolines and Pyrazolo[3,4-b][1,4]diazepines as Potential Antimalarial, Antitrypanosomal and Antileishmanial Agents. Eur J Med Chem 2015; 93: 401-13.
11- Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. Automated Docking Using a Lamarckian Genetic Algorithm and an Empirical Binding Free Energy Function. J Comput Chem 1998; 19(14): 1639-62.
12- Mills N. ChemDraw Ultra 10.0 CambridgeSoft, 100 CambridgePark Drive, Cambridge, MA 02140. Www.cambridgesoft.com. Commercial Price: $1910 for download, $2150 for CD-ROM; Academic Price: $710 for download, $800 for CD-ROM. J Am Chem Soc 2006; 128(41): 13649-50.
13- Jeddi B, Saberi S, Menéndez JC, Sepehri S. Synthesis and Biological Evaluation of Tetrahydropyrimidine and Dihydropyridine Derivatives against Leishmania Major. Acta Parasitol 2022; 67(1): 255-66.
14- Saddala MS, Kumar KK, Rani AU. In Silico Inhibitors for Plasmodium Falciparum Lactate Dehydrogenase. J Bioinforma 2014; 14(2): 146-59.
15- Mishra M, Agarwal S, Dixit A, Mishra VK, Kashaw V, Agrawal RK, et al. Integrated Computational Investigation to Develop Molecular Design of Quinazoline Scaffold as Promising Inhibitors of Plasmodium Lactate Dehydrogenase. J Mol Str 2020; 1207: 127808.
16- Kaushik D, Paliwal D, Kumar A. 2D QSAR and Molecular Docking Studies of Chloroquine Thiazolidinone Derivatives as Potential pfLDH Inhibitors of Plasmodium Falciparum. Int J Pharmacol Pharm Sci 2015; 2(5): 42-53.
17- Shadrack DM, Nyandoro SS, Munissi JJE, Mubofu EB. In Silico Evaluation of Anti-malarial Agents from Hoslundia Opposita as Inhibitors of Plasmodium Falciparum Lactate Dehydrogenase (PfLDH) Enzyme. Comput Mol Biosci 2016; 6(2): 23-32.
18- Zakaria NH, WAI L, Hassan NI. Molecular Docking Study of the Interactions between Plasmodium Falciparum Lactate Dehydrogenase and 4-Aminoquinoline Hybrids. Sains Malays 2020; 49(8): 1905-13.
19- Chaniad P, Mungthin M, Payaka A, Viriyavejakul P, Punsawad C. Antimalarial Properties and Molecular Docking Analysis of Compounds From Dioscorea Bulbifera L. as New Antimalarial Agent Candidates. BMC Complement Med Ther 2021; 21: 144.
20- Shamsuddin MA, Ali AH, Zakaria NH, Mohammat MF, Hamzah AS, Shaameri Z, et al. Synthesis, Molecular Docking, and Antimalarial Activity of Hybrid 4-Aminoquinoline-pyrano[2,3-c]pyrazole Derivatives. Pharmaceuticals 2021; 14(11): 1174.
21- Oluyemi WM, Samuel BB, Adewumi AT, Adekunle YA, Soliman MES, Krenn L. An Allosteric Inhibitory Potential of Triterpenes from Combretum racemosum on the Structural and Functional Dynamics of Plasmodium falciparum Lactate Dehydrogenase Binding Landscape. Chem Biodivers 2022; 19(2): e202100646.
ارسال پیام به نویسنده مسئول
| بازنشر اطلاعات | |
 |
این مقاله تحت شرایط Creative Commons Attribution-NonCommercial 4.0 International License قابل بازنشر است. |
