

دوره 28، شماره 2 - ( اردیبهشت 1399 )
جلد 28 شماره 2 صفحات 2398-2384 |
برگشت به فهرست نسخه ها


![]()
![]()
![]()
Download citation:
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:



BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
Rami M, Fathi M, Rahmati M, Tabandeh M R. Effect of 6 Weeks Endurance Exercise on Hippocampal Pannexin-1 and NLRP-1 Protein Levels in Experimental Diabetic Male Wistar Rats. JSSU 2020; 28 (2) :2384-2398
URL: http://jssu.ssu.ac.ir/article-1-5017-fa.html
URL: http://jssu.ssu.ac.ir/article-1-5017-fa.html
رمی محمد، فتحی محمد، رحمتی مسعود، تابنده محمدرضا. بررسی تأثیر 6 هفته فعالیت استقامتی بر سطوح پروتئینهای Pannexin-1 وNLRP-1 هیپوکمپ در موشهای صحرایی نر ویستار مبتلا به دیابت تجربی. مجله علمي پژوهشي دانشگاه علوم پزشكي شهید صدوقی يزد. 1399; 28 (2) :2384-2398



متن کامل [PDF 610 kb]
(960 دریافت)
| چکیده (HTML) (2538 مشاهده)
متن کامل: (2149 مشاهده)
مقدمه
دیابت ملیتوس (DM) رایجترین اختلال بخش درون ریز پانکراس است که ناشی از کمبود مطلق یا نسبی انسولین و در نتیجه نقص در ترشح این هورمون توسط سلولهای بتا پانکراس میباشد (1). این اختلال که با افزایش مطلق یا نسبی گلوکاگون همراه است، یکی از اختلالات متابولیکی شایع بوده که یکی از علائم اصلی آن هیپرگلیسمی میباشد (2). همزمان با پیشرفت دیابت، هیپرگلیسمی سبب اختلال در سیستمهای قلبی عروقی، کلیه، شبکیه، عدسی چشم، پوست و سیستم عصبی مرکزی و محیطی میشود (3).دیابت به هر دو سیستم عصبی مرکزی و محیطی از طریق آپوپتوز (Apoptosis) نورونهای پایه¬ای آسیب می¬رساند و افزایش قند خون ناشی از دیابت، عوارض میکرو واسکولار شدیدی نظیر نوروپاتی، نفروپاتی و رتینوپاتی (Retinopathy) ایجاد می¬کند. شایع¬ترین عوارض دیابت، نوروپاتی¬های دیابتی هستند که اختلالات سیستم عصبی اتونوم و سیستم عصبی ارادی را بههمراه دارند (4). اختلالات ناشی از آسیب نورونهای هیپوکمپ نیز از عوارض دیابت هستند و نقص در حافظه، یادگیری و شناخت در افراد دیابتی بیشتر از افراد غیر دیابتی گزارش شده است (6,5). هیپوکمپ بهعنوان یک مرکز مهم در حافظه و یادگیری، نسبت به افزایش قند حساس بوده و نورونهای آن در دیابت نوع 1 آسیبپذیر هستند (8, 7). در بیماران دیابتی حجم هیپوکمپ کاهش قابل توجهی را نسبت به بیماران غیر دیابتی نشان میدهد که میتوان دلیل آن را تخریب سلولهای عصبی در این ناحیه که تحت تاثیر بیماری دیابت هستند، عنوان نمود (9). هرچند مکانیسمهای تخریب سلولهای عصبی ناشی از دیابت در هیپوکمپ کاملاً مشخص نشده است. اما مکانیسمهایی نظیر آتروفی دندریتی، تنظیم کاهشی گیرندههای گلوکوکورتیکویید (Glucocorticoids)، تغییر بیان گیرندههای فاکتور رشد شبه انسولینی، کاهش ناقلین انسولین و القای آپوپتوز مطرح شده است. اختلالات عصبی در بیماران مبتلا بهدیابت علاوه بر تغییرات ایسکمیک (Ischemic) قسمت قشری مخ، با افزایش آتروفی (Atrophy) بافت مغز نیز همراه است (10). بهطور ویژه گزارش شده است که قند خون بالا اثرات مخربی بر مناطق ویژه مغز همچون هیپوکمپ داشته و اختلالاتی همچون نقص یادگیری، حافظه، توانائی حل مسئله و همچنین اختلالات ذهنی و حرکتی را در پی دارد (12, 11). نشان داده شده است که مرگ سلولی در دیابت و اختلالات تحلیل برنده مربوط به سیستم عصبی مرکزی (تخریب عصب) متداول است (14, 13). شیوع بیماریهای تخریب عصب در بین بیماران DM رو به افزایش است بهطوریکه در حال حاضر تقریباً 20 درصد از بیماریهای تخریب عصب با DM مرتبط هستند (15). پانکسین (Pannexin-1) یک کانال غشایی است که بهصورت فراوان در CNS و در تمام انواع سلولها مانند میکروگلیا (Microglia)، آستروسیتها (Astrocytes)، اولیگودندروسیتها (oligodendrocyte) و نورونها بیان میشود. بهصورت ویژه، رونوشت Pannexin-1 (Panx-1) در مخچه، شبکیه، قشر مخ، هیپوکمپ، آمیگدال، توده سیاه، پیاز بویایی و نخاع شوکی و دیگر ساختارهای نورونی یافت میشود (16). این پروتئین بهعنوان یک کانال غشایی با قابلیت هدایت غیرانتخابی بالا به مولکولهای کوچک (1.5 kDa) (17)، نفوذپذیری بالا به ATP، Ca+2، گلوتامات و برخی میانجیهای التهابی عمل میکند، و میتواند توسط چندین مکانیسم مانند تحریک مکانیکی، افزایش پتاسیم خارج سلولی، افزایش Ca+2 داخل سلولی و چندین سیگنالینگ درون سلولی فعال شود (18). درک نقش فیزیولوژیکی و پاتولوژیکی Panx-1 بسیار مهم است، زیرا این کانالِ دارای قابلیت هدایتی بالا، دارای ویژگی خاص توانمند سازی برخی گیرندههای لیگاند دار CNS در وضعیتهای پاتولوژیکی مانند التهاب عصبی، دپلاریزاسیون اکسیژن، سکته، مرگ سلولی و تشنج است (19). از سویی تحقیقات نشان داده است که وضعیت هایپرگلیسمیک توسعه یافته در حین DM التهاب پایداری را تولید میکند که میتواند سبب مرگ نورونی شود (21, 20). در سال 2006، تامسون و همکارانش نشان دادند که Panx-1 در پاسخ به محرومیت از اکسیژن و گلوکز (شرایط ایسکمیک) در نورونهای هیپوکمپ ایزوله فعال میشود و یک جریان ثانویۀ شدید را به وجود میآورد که غشاء را دپولاریزه میکند (دپلاریزاسیون اکسیژن)، این شرایط در نهایت منجر به مرگ نورونی میشود (22). اینفلاماسامها (inflammasome) کمپلکسهای چند پروتئینی هستند که مسئول فعال کردن سایتوکاینهای پیش التهابی نظیر اینترلوکین 1 β (IL-1β) و اینترلوکین 18 (IL-18) میباشند (23). شناختهشدهترین اینفلاماسامها، NLRP-1 و NLRP-3 هستند. اینفلاماسامِ NLRP-1 عمدتاً در نورونها وجود دارد (26-24). محققان نشان دادند که بیان NLRP-1 در CNS موشهای دیابتی افزایش مییابد و سبب التهاب عصبی ناشی از مقادیر گلوکز بالا میشود (27). گزارش شده است که استرسهای اکسایشی و همچنین بیماریهای تخریب عصبی نظیر آلزایمر فعالیت NLRP-1 را افزایش داده و سبب بروز التهاب نورونی و تخریب اکسونی میشود (28). فعالیت بدنی فواید سلامتی متعددی نظیر افزایش طول عمر، محافظت در برابر بیماریهای قلبی عروقی، دیابت، سرطان و بیماریهای تخریب عصبی دارد (29). محققان نشان دادند که ورزش سبب توسعه یادگیری و حافظه، تأخیر زوال شناختی مرتبط با سن و کاهش خطر تخریب عصب میشود (30). همچنین تحقیقات نشان داده است که فعالیت بدنی علاوه بر توسعه عملکرد رفتاری، سبب ارتقاء شکلپذیری سیناپسی در هیپوکمپ، که یک ساختار کلیدی برای یادگیری است، میشود (31). علاوه بر این، فعالیت بدنی میتواند سطوح سایتوکاینهای پیش التهابی را در مغز کاهش دهد (32). بنابراین، کاهش التهاب مرکزی و محیطی توسط فعالیت بدنی میتواند بهعنوان مکانیزمی متداول برای کاهش خطر هم دیابت و هم زوال شناختی به خدمت گرفته شود (30). در پژوهشهای انجام شده در زمینه فعالیت بدنی و ورزش، گزارش شده است که انجام تمرین استقامتی میتواند دارای اثرات ضداکسایشی نیز باشد و از طریق افزایش میزان آنزیمهای ضداکسایشی، بیماران DM را در مقابل فشار اکسایشی محافظت کند (33). تحقیقات اخیر نشان داده که آسیب اکسایشی در هیپوکمپ هنگام فعالیت با شدت زیر بیشینه کاهش یافته و ورزش منظم میتواند با اثر ضداکسایشی عملکرد حافظه را ارتقاء دهد (35, 34). هرچند ارتباط بیماری DM با اختلالات سیستم عصبی مرکزی و اثرات تخریبی آن بر بافتهای عصبی تا حدودی گزارششده است، با اینحال سازوکار درگیر آن بهخوبی مشخص نیست (37, 36). با توجه به اهمیت اختلالات تخریب عصب در اعمال طبیعی مغز و همچنین اختلالات گزارششده در عملکرد هیپوکمپ بیماران DM، در پژوهش حاضر سعی بر آن است که ارتباط اثر ضدالتهابی و ضداکسایشی فعالیت ورزشی به صورت استقامتی با تغییرات سطوح Panx-1 و پروتئین NLRP-1 بررسی شود. همچنین مطالعه حاضر بهدنبال پاسخگوئی به این سوال است که آیا تمرینات استقامتی قادر است بهعنوان یک راهبرد غیرداروئی این تغییرات احتمالی را تعدیل کند؟ از این رو در مطالعه حاضر، به بررسی تاثیر تمرین استقامتی بر میزان پروتئینهای Panx-1 و NLRP-1 بافت هیپوکمپ موشهای صحرائی نر ویستار دارای دیابت پرداختیم.
روش بررسی
پژوهش حاضر از نوع تجربی و بهشیوه آزمایشگاهی و با طرح پس آزمون آزمون بههمراه گروه کنترل است. جهت انجام آزمایش،32 سر موش صحرایی نر ویستار با 10 هفته سن و میانگین توده بدنی9/4±245 گرم بهعنوان نمونه تحقیق از مرکز نگهداری حیوانات دانشگاه علوم پزشکی لرستان خریداری شد. پس از آشنایی با محیط آزمایشگاه و نوار گردان، 18 سر از رتها از طریق تزریق درون صفاقی STZ (Streptozotocin) دیابتی شده و پس از تأیید القای دیابت و متحمل شدن تلفاتی به تعداد 4 سر رت در 24 ساعت پس از تزریق، 28 رت باقیمانده به روش تصادفی به چهار گروه بدین شرح تقسیم شدند: 1)گروه دیابتی تمرین (DT): این گروه شامل 7 سر رت دیابتی شده بودو از هفته دوازدهم زندگی به مدت6 هفته و هر هفته 5 جلسه تمرین استقامتی انجام میدادند و پس از آخرین جلسه تمرینی تشریح شدند. 2) گروه دیابتی کنترل (DC): این گروه شامل 7 سر رت دیابتی شده بود و در هیچگونه برنامه تمرینی شرکت داده نشدند. این رتها همزمان با بقیه گروهها تشریح شده و کلیه مراحل و آزمایشها مطابق دیگر گروهها بر روی آنها انجام پذیرفت. 3) گروه تمرینی سالم (HT): این گروه شامل 7رت بود که همانند گروه DT در برنامه تمرینی نوارگردان شرکت داده شدند. 4) گروه کنترل سالم (HC): این گروه شامل 7 رت بود که درگیر هیچ فعالیتی نبودند. این رتها (گروه 3 و 4) نیز همزمان با بقیه گروهها تشریح شده و کلیه مراحل و آزمایشها مطابق دیگر گروهها بر روی آنها انجام گرفت. کلیه رتها در شرایط کنترل شده محیطی با میانگین دمای 3±22 درجه سانتیگراد، چرخه روشنایی-تاریکی 12:12 ساعت (شروع چرخه بیداری ساعت 16) و با دسترسی آزاد به آب و غذای ویژه موش نگهداری گردیدند. حیوانات به مدت 2 هفته با شرایط آزمایشگاه و نوار گردان مخصوص جوندگان آشنا شدند. در طول مرحله آشنا سازی، بهمنظور آشنا شدن با شرایط آزمایشگاه، نوار گردان و دستکاری، حیوانات 5 روز در هفته به مدت 15-10 دقیقه با سرعت 10 متر در دقیقه بر روی نوار گردان راه رفتند. تمام جلسات تمرینی در پایان سیکل خواب حیوانات و بین ساعت های 16 تا 18 عصر برگزار گردید. لازم به ذکر است که بهمنظور انجام آزمایشات مولکولی تعداد 5 نمونه از هر گروه مورد سنجش قرار گرفت. موشها در اطاقی به ابعاد 2 متر در 7 متر در شرایط کنترل شده نور (12 ساعت روشنایی و 12 ساعت تاریکی)، دما (3±22 درجه سانتیگراد)، و رطوبت (حدود 45 درصد) نگهداری شدند. تعداد 4 تا 6 عدد موش در قفسهایی از جنس پلکسی گلاس با درب توری نگهداری شدند که آزادانه به آب و غذای استاندارد دسترسی داشتند. پس از 12 ساعت محرومیت از غذا، با تزریق درون صفاقی mg/kg 45محلولSTZ ((Sigma, St. Louis MO, USA، تهیه شده در بافر سیترات تازه 0/5 مولار با pH=4/5) دیابت القاء گردید. بهصورتی که به ازای هر کیلوگرم موش ml2 بافر سیترات به 45 میلیگرم STZ اضافه می شد و با تناسب بستن بین وزن نهایی موش و مقدار بافر سیترات و STZ، حجم نهایی محلول STZ به منظور تزریق مشخص میشد. به رتهای غیر دیابتی نیز معادل حجمی بافر سیترات تزریق گردید. 48 ساعت پس از تزریق، با ایجاد یک جراحت کوچک توسط لانست روی ورید دم رتها، یک قطره خون روی نوار گلوکومتر قرار داده شد و قند خون با استفاده از دستگاه گلوکومتری (Roche Diagnostics K.K., Tokyo, Japan) قرائت گردید. رتهایی که قند خون آنها بالاتر از 300 mg/dl بود، به عنوان رتهای دیابتی در مطالعه حاضر مورد استفاده قرار گرفتند (38). در پژوهش حاضر جهت انجام یک دوره فعالیت استقامتی از پروتکل مطالعه چاء و همکاران (39) استفاده شد؛ بدینصورت که گروههای ورزشی در معرض تمرین نوار گردان برای 5 جلسه در هفته و به مدت 6 هفته قرار گرفتند. سرعت و مدت تمرین نوار گردان بهتدریج افزایش یافت و از 10 متر در دقیقه برای 10 دقیقه در هفته اول، 10 متر در دقیقه برای 20 دقیقه در هفته دوم، 15 متر در دقیقه برای 20 دقیقه در هفته سوم، 15 متر در دقیقه برای 30 دقیقه در هفته چهارم، به 18-17 متر در دقیقه برای 30 دقیقه در هفته پنجم افزایش یافت. جهت رسیدن به سازگاری های بهدست آمده درحالت یکنواخت، تمامی متغیرهای تمرینی درهفته پایانی (هفته ششم) ثابت نگهداشته شدند (جدول1). در پایان 6 هفته برنامه تمرینی، 24 ساعت پس از آخرین جلسه تمرین، رتها توسط تزریق درون صفاقی ترکیب کتامین (mg/kg-175) و زایلازین (mg/kg-1 5) بیهوش شده و پس از جدا کردن سر توسط گیوتین و تحت شرایط استریل بافت هیپوکمپ جدا شده و بلافاصله به فریزر 70- (88FD-2-93-A، ساخت ایران) منتقل شد. ارزیابی بیان پروتئین های Pannexin-1و NLRP-1با استفاده از روش وسترن بلات صورت گرفت. به این صورت که نمونههای بافت هیپوکمپ ابتدا در لایزیس بافر هموژن شدند (لایزیس بافر شامل: Tris-Hcl (0/3 گرم، 50 میلیمول در لیتر) ، تریتونX-100 (0/02گرم، 0/1٪)،کلسیم دی اکسید سدیم (0/05 گرم،0/25٪)،NaCl (0/43 گرم، 150 میلیمول در لیتر)، SDS (0/02 گرم،0/1٪)، اسید اتیلن دی آمین تراستیک (EDTA ، 5/84 گرم) که در 20 میلیلیتر آب مقطر و در7/4=pH مخلوط شده و هموژن شدند (16000 rpm و بهمدت 20 دقیقه در 4 درجهسانتیگراد). برای هر 10 میلیلیتر (10X) از یک قرص مهارکننده پروتئاز استفاده شد. غلظت سوپرنیتان بهدست آمده توسط کیت بردفورد مورد بررسی قرار گرفت. پروتئینها با استفاده از الکتروفورز بر روی ژل SDS-PAGE (10٪) بارگذاری شدند (ابتدا در ولتاژ 60 به مدت 15 دقیقه و سپس در ولتاژ 100 به مدت یک ساعت نگهداری شد). سپس پروتئینها طی فرآیند انتقال بهمدت 105 دقیقه روی کاغذ نیتروسلولوز انتقال داده شدند و سپس با آنتیبادیهای اولیه مونوکلونال Pannexin-1(Human/Mouse/Rat Pannexin-1 Antibody (MAB7097):R&D system.)، NLRP-1(Rat, Human Anti-NALP1 antibody (ab3683): abcam. 100ug) و β-actin (Anti-beta Actin antibody [mabcam 8226]) مورد آزمایش قرار گرفتند (رقت 1: 2000 در PBS). از محلول 5 درصد اسکیم میلک برای مسدود کردن غشا استفاده شد (یک شب) و از آنتیبادی ثانویه (AS09 618 Goat anti-Rat IgG (H&L), HRP conjugated, Agrisera Sweden) نیز برای مدت یک ساعت برای اتصال به آنتیبادی اولیه استفاده شد. باندهای پروتئین در دستگاه Bio-Rad Gel Doc با استفاده از کیت ECL ظاهر شدند. سپس نوارهای پروتئینی با استفاده از نرمافزار Image J در مرحله آخر روش وسترن بلات مورد بررسی قرار گرفت.
تجزیه و تحلیل آماری
ابتدا نرمال بودن توزیع دادهها با استفاده از آزمون شاپیروویلک ارزیابی شد و مشخص شد که توزیع دادهها طبیعی میباشند. در ادامه از آزمون لون (Levene’s Test) برای تعیین همگن بودن واریانسها استفاده شد. برای مقایسه گروهها در متغیرهای مورد مطالعه از تحلیل واریانس یکطرفه استفاده شد. از آزمون توکی بهعنوان آزمون تعقیبی استفاده شد. سطح معناداری نیز P≤ 0/05 در نظر گرفته شد. کلیه بررسیهای آماری با استفاده از نرم افزار version 18 SPSS انجام گرفت.
ملاحظات اخلاقی
پروپوزال این تحقیق توسط کمیته اخلاق دانشگاه لرستان با کد کمیته اخلاق پژوهش بر حیوانات به شماره LU.ECRA.2017.2 مورد بررسی و تایید قرار گرفت.
نتایج
تغییرات وزن بدن و گلوکز خون موشها در گروه¬های مختلف آزمایشی در طی دوره تمرین در شکل 1 و 2 نشان داده شده است. همان¬گونه که در شکل 1 نشان داده شده است وزن اولیه گروهها اختلاف معناداری با یکدیگر نداشت (P>0/05)، اما در پایان پژوهش، میانگین تغییرات وزن موشهای گروه کنترل دیابت نسبت به کنترل سالم و نیز گروه دیابتی تمرین کرده نسبت به گروه کنترل سالم به صورت معناداری کمتر بود (به ترتیب P=0/004 و P=0/003). میانگین تغییرات وزن گروه دیابت تمرین کرده نسبت به گروه سالم تمرین کرده بهصورت معناداری کمتر بود (P=0/003). نتایج تحلیلهای آماری نشان داد که میانگین تغییرات وزن گروههای تمرین کرده سالم نسبت به کنترل سالم معنادار نبود (P=0/09). همچنین، میانگین تغییرات وزن گروههای دیابت تمرین کرده نسبت به کنترل دیابت اگرچه پس از شش هفته تمرین افزایش یافت، اما این افزایش معنادار نبود (P>0/05). همانگونه که در شکل 2 نشان داده شده است در شروع برنامه تمرینی سطح گلوکز خون بهصورت معناداری 48 ساعت پس از القای دیابت توسط استرپتوزوتوسین در موشهای گروه های دیابتی افزایش یافت (P<0/001)، و پس از 6 هفته تمرین استقامتی در مقایسه با گروههای سالم همچنان از اختلاف معنیداری برخوردار بود (P<0/001)، همچنین، در پایان برنامه تمرینی، غلظت گلوکز خون گروه دیابت تمرین کرده نسبت به گروه دیابت کنترل به صورت معناداری پایینتر بود (P<0/001). تغییرات سطوح بیان پروتئینهای Pannexin-1 و NLRP-1، پس از انتهای دوره تمرینی در جدول 2 نشان داده شده است.
شکل3 میزان بیان پروتئین Pannexin-1 را نسبت به β-actin در گروههای مختلف نشان میدهد.
در شکل 4 نمونههایی از روند تغییرات بیان پروتئین Panx-1 در بافت هیپوکمپ گروههای مختلف، که با روش وسترن بلات بر روی کاغذ نیتروسلولز مشخص (Detect) شدهاند، نشان داده شده است. در این تحقیق از سطوح بیان پروتئین β-actin بهعنوان کالیبراتور استفاده شده و تغییرات بیان پروتئینهای مورد نظر با توجه به بیان ثابت این پروتئین ارزیابی گردید. برای کمیسازی باندهای پروتئینی، دانسیته آنها توسط نرمافزار Image J محاسبه شد. نتایج نشان دادکه بیماری دیابت موجب افزایش معنادار سطوح بیان پروتئین Panx-1 در مقایسه با موشهای سالم شد (P<0/05). همچنین یک دوره تمرین استقامتی سطوح بیان این پروتئین را در موشهای دیابتی کاهش داده است (P<0/05). بهعلاوه تمرین استقامتی در موشهای دیابتی سطوح بیان پروتئین Panx-1 تا حدی کاهش داده است که با سطوح این پروتئین در گروه کنترل سالم اختلاف معناداری ندارد (P>0/05).
شکل 5 میزان بیان پروتئین NLRP-1 را نسبت به β-actin در گروه های مختلف نشان میدهد. نتایج بیانگر این نکته است که بیماری دیابت موجب افزایش معنادار سطوح بیان پروتئین NLRP-1 در مقایسه با موش های سالم است (P<0/05). نتایج همچنین نشان داد که یک دوره تمرین استقامتی سطوح این پروتئین را در موشهای دیابتی کاهش داده است (P<0/05). در شکل 6 نمونه هایی از روند تغییرات بیان پروتئین NLRP-1 در بافت هیپوکمپ گروههای مختلف، که با روش وسترن بلات بر روی کاغذ نیتروسلولز مشخص (Detect) شدهاند، نشان داده شده است.
جدول1: نمایش عددی پروتکل در هفتههای مختلف.

جدول 2: میانگین و انحراف معیار تغییرات سطوح بیان پروتئینهای Panx-1 و NLRP-1 در موشهای گروههای مختلف

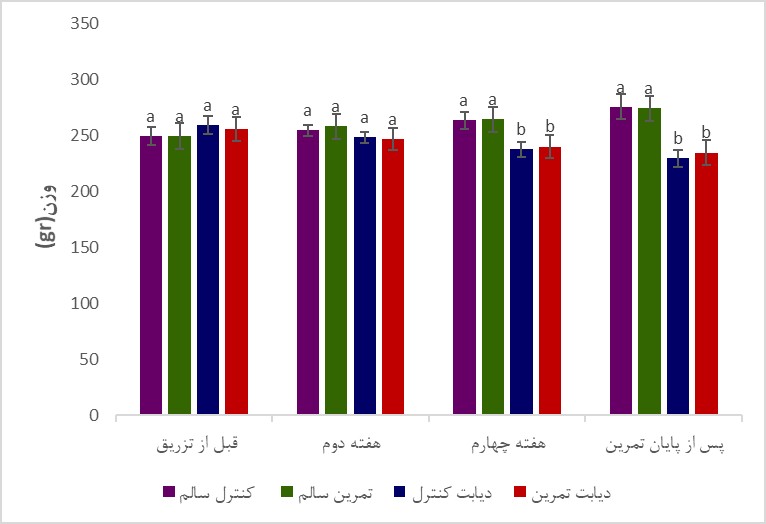
شکل 1: تغییرات میانگین وزن بدن در موش های گروههای مختلف.
حروف نامتشابه بیانگر وجود اختلاف آماری معنیدار در بین گروهها میباشد (P<0/05).
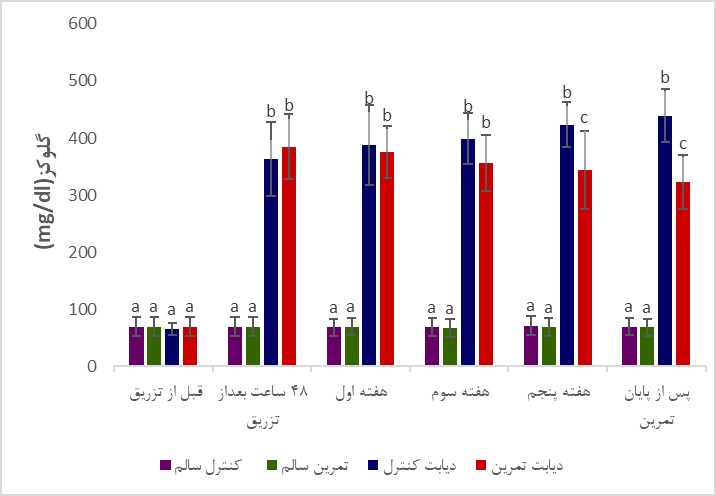
شکل 2: تغییرات میانگین سطح گلوکز خون در موشهای گروههای مختلف.
حروف نامتشابه بیانگر وجود اختلاف آماری معنیدار در بین گروهها میباشد (P<0/05).

شکل 3: میزان بیان پروتئین Pannexin-1در موش های گروه های مختلف.
حروف نامتشابه بیانگر وجود اختلاف آماری معنیدار در بین گروهها میباشد (P<0/05).
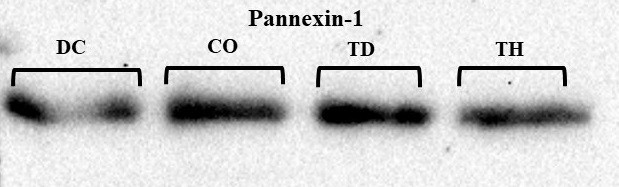

شکل 4 :باندهای وسترنبلات پروتئین Pannexin-1 در بافت هیپوکمپ موشهای گروههای مختلف.
به ترتیب از چپ به راست: DC) گروه دیابت کنترل؛ CO) گروه کنترل سالم؛ TD) گروه دیابت تمرین؛ TH) گروه تمرین سالم.
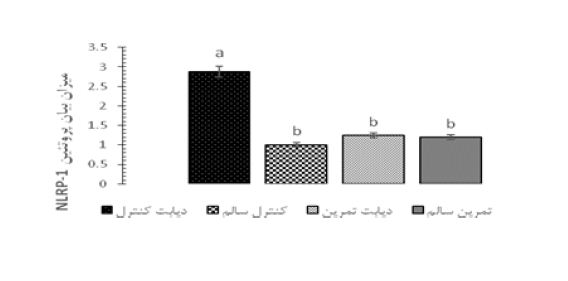
شکل 5: میزان بیان پروتئین NLRP-1 در موشهای گروههای مختلف.
حروف نامتشابه بیانگر وجود اختلاف آماری معنیدار در بین گروهها میباشد (P<0/05).


شکل 6: مقایسه سطوح بیان پروتئین NLRP-1در بافت هیپوکمپ موشهای گروههای مختلف.
به ترتیب از چپ به راست: DC) گروه دیابت کنترل؛ CO) گروه کنترل سالم؛ TD) گروه دیابت تمرین؛ TH) گروه تمرین سالم
بحث
پژوهش حاضر با هدف بررسی تأثیر یک دوره فعالیت استقامتی بر سطوح بیان پروتئینهای Panx-1 و NLRP-1 در بافت هیپوکمپ موش¬های صحرایینر ویستار دارای دیابت انجام شد. نتایج حاصل از سنجش منظم مقادیر گلوکز خون در طی این تحقیق نشان داد که یک دوره فعالیت بدنی کاهش پایداری را در سطوح گلوکز خون در موشهای صحرایی دیابتی ایجاد میکند. انجمن دیابت آمریکا در سال 2006 به بررسی اثر فعالیت بدنی بر سطح گلوکز پلاسما افراد دیابتی پرداخت. آنها گزارش کردند که ورزش هوازی طولانیمدت بهصورت پایداری سطح گلوکز پلاسما را در افراد مبتلا به دیابت نوع 1 کاهش میدهد (40)، همچنین گوئلفی و همکارانش (2005) نیز در مطالعه خود بیان کردند که هم فعالیت تناوبی با شدت بالا و هم فعالیت با شدت متوسط سطوح گلوکز خون را، در حین تمرین و در زمان بازگشت به حالت اولیه، در افراد مبتلا به دیابت نوع 1 کاهش میدهد (41). فعالیت بدنی پاسخهای هورمون رشد و کاتکولامینها را تحریک کرده و از سقوط گلوکز در حین فعالیت جلوگیری میکند. از سوی دیگر، این امر نشان دهنده این واقعیت است که فعالیت بدنی در زمانی که افزایش تولید گلوکز درون زاد مورد نیاز جبران نمیشود، سبب افزایش بهره برداری از گلوکز میشود (40). نتایج تحقیق حاضر نشان داد که بیماری دیابت منجر به افزایش معنادار سطوح بیان پروتئین Panx-1 در بافت هیپوکمپ موشهای صحرایی نر میشود. نتایج تحقیقات فانگ منگ در سال 2013 نیز نشان داد که بیان Panx-1تحت شرایط هایپرگلیسمیک افزایش مییابد. بنابراین، از آنجا که افزایش بیان این پروتئین سبب راه اندازی آبشارهای سیگنالینگ تولید شاخصهای التهابی میشود، مهار Panx-1 از رهایش فاکتورهای التهابی و آسیبنورونی ناشی از هایپرگلیسمی جلوگیری میکند. از سویی دیگر، علاوه بر نقش ROS (Reactive Oxygen Species)، بهویژه نیتریک اکساید (NO)، در افزایش بازشدن همیکانال Panx-1، ROS در غلظتهای بالا بهعنوان مولکولهای سمی منجر به آسیب بافتی و سپس تغییر یا تخریب انواع سلولها میشود (27). غلظت بالای گلوکز خون در دراز مدت سبب آسیب بافتی می¬شود. اگر گلوکز خون در دیابت مدت زیادی کنترل نشود، عروق خونی بافت¬های متعدد بدن دچار اختلال شده و دستخوش تغییرات ساختمانی می¬شوند که موجب عدم خونرسانی کافی به بافت¬ها می¬گردد. بدین ترتیب خطر حملات قلبی، سکته مغزی، بیماری مرحله نهایی کلیه، رتینوپاتی و کوری، ایسکمی و گانگرن اندام¬ها افزایش می¬یابد (42, 2, 1). این مسئله بسیار مهم است که شرایط ایسکمیک همیکانالهای Panx-1 را در نورونها باز میکند، در واقع تحقیقات نشان داده شده است که رادیکالهای آزاد تولید شده ناشی از شرایط ایسکمیک، از جمله NO، در باز شدن همیکانال Panx-1 نورونی درگیر هستند (43). سایتوکین ها و فاکتورهای رشد فرآیندهای فیزیولوژیکی و پاتولوژیکی متعددی را در سیستم عصبی مرکزی تنظیم میکنند (44). تعدادی از مطالعات نشان میدهند که همیکانالها همچنین توسط سایتوکینها و فاکتورهای رشد تنظیم میشوند. عوامل پیش التهابی مانند TNF-α، IL-1βو پپتید آمیلوئید β منجر به رهایش ATP از طریق همیکانالهای Pannexin و Connexin در آستروسیتها و میکروگلیا میشود (46, 45) و سبب انتشار موج کلسیم به واسطه فعالیت گیرندههای P2 در سلولهای گلیال میشود. از دیگر نتایج این پژوهش می توان به افزایش معنادار سطوح بیان پروتئین NLRP-1 در بافت هیپوکمپ موشهای صحرایینر مبتلا به بیماری دیابت اشاره کرد. این یافته با نتایج فانگ منگ و همکارانش در سال 2014 همسو است. آنها در تحقیق خود بیان کردند که بیان اینفلاماسامِ NLRP-1 در ناحیه کورتکس مغز تحت شرایط هایپرگلیسمیک ناشی از بیماری دیابت تنظیم مثبت شده و به آسیب نورونی منجر شده است (27). فعالیت اینفلاماسامها به عنوان یک ماشین مولکولی درون سلولی برای شروع آسیب نورونی عمل کرده و سرانجام منجر به اختلال عملکرد نورون و آپوپتوزیس می شود (27). این نتایج نشان میدهد که فعالیت اینفلاماسامِ NLRP-1 ممکن است مکانیسم اصلی آغاز آسیب نورونی در شرایط هایپرگلیسمیک باشد. فانگ منگ و همکارانش در تحقیق خود بیان کردند که همیکانال Panx-1 فعالیت اینفلاماسامِ NLRP-1 را در نورون میانجیگری میکند. در واقع، مهار همیکانال Panx-1، التهاب نورونی را در حین شرایط هایپرگلیسمی تضعیف میکند. این یافتهها نشان داد که اینفلاماسامِ NLRP-1 مکانیسم مهمی برای راه اندازی پاسخهای التهابی موضعی در نورونها است (27). نتایج حاصل از تحقیق حاضر نشان داد که یک دوره فعالیت استقامتی منجر به تعدیل سطوح بیان پروتئین های Panx-1 و NLRP-1 و نزدیک ساختن آنها به سطح نرمال، در بافت هیپوکمپ موشهای گروه دیابت تمرین گردید. ورزش و فعالیت بدنی بهعنوان کاهش دهنده پاسخ التهابی سیستمیک مزمن شناخته شده است و اثرات آنتی اکسیدانی و نیز اثرات مثبتی بر شکل پذیری سیناپسی جوندگان دیابتی و یا چاق نشان داده است (47, 30). اسپیک و همکارانش در سال 2014 بهبررسی اثر ضداکسایشی فعالیت ورزشی بر هیپوکمپ پرداختند. آنها نشان داده که آسیب اکسایشی در هیپوکمپ هنگام فعالیت با شدت زیر بیشینه کاهش یافته و ورزش منظم میتواند با اثر ضداکسایشی عملکرد حافظه را ارتقاء دهد (34). بالدوسی و همکارانش به بررسی اثر ضد التهابی فعالیت ورزشی بر روی افراد دیابتی پرداخته و نتایج پژوهش آنها نشان داد که فعالیت بدنی منظم میتواند عوامل التهابی نظیر TNF-α را در جریان گردش خون این بیماران کاهش دهد، همچنیننشان داد که فعالیت بدنی سبب کاهش ROS و افزایش سطوح دفاع ضداکسایشی میشود (48). همچنین چیریکو و همکارانش در سال 2016 کاهش معنیداری را در IL-1β (سایتوکین پردازش شده توسط اینفلاماسامها) در مغز موشهای تیمار شده با رژیم پرچرب، پس از وادار کردن حیوان به تمرین استقامتی، نشان دادند (49). در هیپوکمپ موشهای دیابتی شده با STZ، نه تنها افزایش بارز گونههای اکسیژن فعال مشاهده شده است، بلکه همچنین فعالیت پایدار NF-KB قابل مشاهده است (51, 50). فعال شدن NF-KBمیتواند منجر به تولید محصولات سایتوکسیک شود که التهاب و استرس اکسیداتیو را تشدید کرده و باعث اختلال در عملکرد سلول و متعاقب آن افزایش آپوپتوزیس یا مرگ سلولی میشود (52). از سویی بهخوبی نشان داده شده است که در حین فرآیندهای التهابی در بیماریهای تخریب عصب نظیر دیابت، باز شدن همیکانالها ایمنی سلولهای عصبی را کاهش میدهد (44). همانگونه که در بیماریهایی نظیر دیابت میلیتوس مشاهده میشود فرآیندهای تخریب عصب، که با التهاب عصبی همراه هستند، ممکن است باعث افزایش فعالیت همیکانالهای آستروگلیال و نورونی شود، که منجر به مرگ سلول و تخریب عملکرد دستگاه عصبی مرکزی میشود (53). از سوی دیگر در وضعیت پاتولوژیکی نظیر شرایط ایسکمیک، جایی که رادیکالهای آزاد به ویژه NO افزایش مییابند، همیکانالهای غشایی Panx-1 در حالت باز شده قرار دارند (44) و آبشار سیگنالینگی را راه اندازی میکنند که نهایتاً منجر به مرگ سلولی میشود (53). علاوه بر این که باز شدن همیکانالهای غشایی خود عامل تحریکی برای فعال شدن اینفلاماسامها است، اتصال ATP ترشح شده از سلولهای التهابی به کانالهای P2X، جریان یونی Na+، Ca2+ و K+را در عرض منافذ به راه میاندازد (ورود بیشتر Ca2+ فعالیت آبشارهای نوروتوکسیک درون سلولی را افزایش میدهد) که کمپلکس اینفلاماسام-کاسپاز-1 را فعال کرده و سبب شکسته شدن سایتوکینهای پیشالتهابی به اشکال فعال آنها مانند IL-1β و IL-18 میشود، که قادر هستند نوعی از مرگ سلولی برنامهریزی شده که با نام پریوپتوزیس (خودکشی سلولی) شناخته شده است را میانجیگری کنند (54). بنابراین، سازگاری ناشی از فعالیت بدنی با شدت متوسط و یا با شدت زیر بیشینه احتمالاً میتواند با کاهش رهایش شاخصهای التهابی و نیز کاهش رادیکالهای آزاد و آسیبهای اکسایشی، فعالیت همیکانالهای غشایی مانند Panx-1 و متعاقب آن فعالیت اینفلاماسامهایی نظیر NLRP-1 را مهار کند. باید خاطر نشان کرد که علاوه بر این که عواملی نظیر شاخصهای التهابی بیان این دو پروتئین را افزایش میدهند، افزایش بیان این دو پروتئین بهصورت چرخهای معیوب، خود سبب افزایش شاخصهای التهابی و تسریع فرآیند مرگ سلولی میشوند (55). علاوه بر فواید قلبی عروقی فعالیت بدنی و همپوشانی آنها با سلامت مغز، اثرات فعالیت بدنی بر جلوگیری از ناهنجاریهای تخریب عصب و گسترش بی رویه دارو درمانی نیز میتواند جالب توجه باشد (56). نشان داده شده است که Panx-1 در پاسخ به محرومیت از اکسیژن و گلوکز (شرایط ایسکمیک) در نورونهای هیپوکمپ یک جریان ثانویۀ شدید را بهوجود میآورد که غشاء را دپولاریزه میکند (دپلاریزاسیون اکسیژن)، این شرایط در نهایت منجر به مرگ نورونی میشود (22). افزایش شبکه عروقی و جریان خون ناشی از فعالیت بدنی در بیماران دیابتی ممکن است سبب در دسترس قرار گرفتن بیشتر اکسیژن در بافت مغزی شود. در نتیجه، احتمالاً در دسترس قرار داشتن اکسیژن سبب مهار بیان بیش از اندازه این همیکانال Panx-1شده و از راهاندازی چرخه معیوب بیان کمپلکس پروتئینی NLRP-1 و شاخصهای التهابی در سلولهای آستروگلیال و نورونهای بافت هیپوکمپ موشهای صحرایی دیابتی جلوگیری میکند. در این تحقیق تنها بیان این پروتئینها در بافت هیپوکمپ مورد ارزیابی قرار گرفته است، مطالعات بیشتری نیاز است تا عوامل گوناگون موثر در بیان چنین پروتئینهایی را در اجزای مختلف سیستم عصبی مرکزی مورد بررسی قرار دهد.
نتیجهگیری
نتایج این پژوهش نشان داد که فعالیت استقامتی با شدت متوسط و یا زیر بیشینه میتواند مهار کننده خوبی برای کنترل بیان بیش از اندازه پروتئینهای Panx-1 و NLRP-1 در بافت هیپوکمپ موشهای دیابتی باشد. چنین مهاری میتواند از راهاندازی چرخههای معیوب درون سلولی جلوگیری کرده و به بیان دیگر شرایط تحریک کننده تخریب عصبی را کنترل کند.
سپاسگزاری
این پژوهش حاصل رساله دکتری رشته فیزیولوژی ورزشی (گرایش عصبی- عضلانی) دانشگاه لرستان است که در تاریخ 27/11/1395 و با شماره 968/تگ به تصویب رسیده است. بدین وسیله از معاونت پژوهشی دانشگاه لرستان و همکاری دانشگاه شهید چمران اهواز تشکر و قدردانی میشود. هزینه اجرای این پژوهش از محل اعتبارات پژوهانه واحد پژوهشی دانشگاه لرستان تامین شده است.
حامی مالی: معاونت محترم پژوهشی دانشگاه لرستان.
تعارض در منافع: وجود ندارد.
دیابت ملیتوس (DM) رایجترین اختلال بخش درون ریز پانکراس است که ناشی از کمبود مطلق یا نسبی انسولین و در نتیجه نقص در ترشح این هورمون توسط سلولهای بتا پانکراس میباشد (1). این اختلال که با افزایش مطلق یا نسبی گلوکاگون همراه است، یکی از اختلالات متابولیکی شایع بوده که یکی از علائم اصلی آن هیپرگلیسمی میباشد (2). همزمان با پیشرفت دیابت، هیپرگلیسمی سبب اختلال در سیستمهای قلبی عروقی، کلیه، شبکیه، عدسی چشم، پوست و سیستم عصبی مرکزی و محیطی میشود (3).دیابت به هر دو سیستم عصبی مرکزی و محیطی از طریق آپوپتوز (Apoptosis) نورونهای پایه¬ای آسیب می¬رساند و افزایش قند خون ناشی از دیابت، عوارض میکرو واسکولار شدیدی نظیر نوروپاتی، نفروپاتی و رتینوپاتی (Retinopathy) ایجاد می¬کند. شایع¬ترین عوارض دیابت، نوروپاتی¬های دیابتی هستند که اختلالات سیستم عصبی اتونوم و سیستم عصبی ارادی را بههمراه دارند (4). اختلالات ناشی از آسیب نورونهای هیپوکمپ نیز از عوارض دیابت هستند و نقص در حافظه، یادگیری و شناخت در افراد دیابتی بیشتر از افراد غیر دیابتی گزارش شده است (6,5). هیپوکمپ بهعنوان یک مرکز مهم در حافظه و یادگیری، نسبت به افزایش قند حساس بوده و نورونهای آن در دیابت نوع 1 آسیبپذیر هستند (8, 7). در بیماران دیابتی حجم هیپوکمپ کاهش قابل توجهی را نسبت به بیماران غیر دیابتی نشان میدهد که میتوان دلیل آن را تخریب سلولهای عصبی در این ناحیه که تحت تاثیر بیماری دیابت هستند، عنوان نمود (9). هرچند مکانیسمهای تخریب سلولهای عصبی ناشی از دیابت در هیپوکمپ کاملاً مشخص نشده است. اما مکانیسمهایی نظیر آتروفی دندریتی، تنظیم کاهشی گیرندههای گلوکوکورتیکویید (Glucocorticoids)، تغییر بیان گیرندههای فاکتور رشد شبه انسولینی، کاهش ناقلین انسولین و القای آپوپتوز مطرح شده است. اختلالات عصبی در بیماران مبتلا بهدیابت علاوه بر تغییرات ایسکمیک (Ischemic) قسمت قشری مخ، با افزایش آتروفی (Atrophy) بافت مغز نیز همراه است (10). بهطور ویژه گزارش شده است که قند خون بالا اثرات مخربی بر مناطق ویژه مغز همچون هیپوکمپ داشته و اختلالاتی همچون نقص یادگیری، حافظه، توانائی حل مسئله و همچنین اختلالات ذهنی و حرکتی را در پی دارد (12, 11). نشان داده شده است که مرگ سلولی در دیابت و اختلالات تحلیل برنده مربوط به سیستم عصبی مرکزی (تخریب عصب) متداول است (14, 13). شیوع بیماریهای تخریب عصب در بین بیماران DM رو به افزایش است بهطوریکه در حال حاضر تقریباً 20 درصد از بیماریهای تخریب عصب با DM مرتبط هستند (15). پانکسین (Pannexin-1) یک کانال غشایی است که بهصورت فراوان در CNS و در تمام انواع سلولها مانند میکروگلیا (Microglia)، آستروسیتها (Astrocytes)، اولیگودندروسیتها (oligodendrocyte) و نورونها بیان میشود. بهصورت ویژه، رونوشت Pannexin-1 (Panx-1) در مخچه، شبکیه، قشر مخ، هیپوکمپ، آمیگدال، توده سیاه، پیاز بویایی و نخاع شوکی و دیگر ساختارهای نورونی یافت میشود (16). این پروتئین بهعنوان یک کانال غشایی با قابلیت هدایت غیرانتخابی بالا به مولکولهای کوچک (1.5 kDa) (17)، نفوذپذیری بالا به ATP، Ca+2، گلوتامات و برخی میانجیهای التهابی عمل میکند، و میتواند توسط چندین مکانیسم مانند تحریک مکانیکی، افزایش پتاسیم خارج سلولی، افزایش Ca+2 داخل سلولی و چندین سیگنالینگ درون سلولی فعال شود (18). درک نقش فیزیولوژیکی و پاتولوژیکی Panx-1 بسیار مهم است، زیرا این کانالِ دارای قابلیت هدایتی بالا، دارای ویژگی خاص توانمند سازی برخی گیرندههای لیگاند دار CNS در وضعیتهای پاتولوژیکی مانند التهاب عصبی، دپلاریزاسیون اکسیژن، سکته، مرگ سلولی و تشنج است (19). از سویی تحقیقات نشان داده است که وضعیت هایپرگلیسمیک توسعه یافته در حین DM التهاب پایداری را تولید میکند که میتواند سبب مرگ نورونی شود (21, 20). در سال 2006، تامسون و همکارانش نشان دادند که Panx-1 در پاسخ به محرومیت از اکسیژن و گلوکز (شرایط ایسکمیک) در نورونهای هیپوکمپ ایزوله فعال میشود و یک جریان ثانویۀ شدید را به وجود میآورد که غشاء را دپولاریزه میکند (دپلاریزاسیون اکسیژن)، این شرایط در نهایت منجر به مرگ نورونی میشود (22). اینفلاماسامها (inflammasome) کمپلکسهای چند پروتئینی هستند که مسئول فعال کردن سایتوکاینهای پیش التهابی نظیر اینترلوکین 1 β (IL-1β) و اینترلوکین 18 (IL-18) میباشند (23). شناختهشدهترین اینفلاماسامها، NLRP-1 و NLRP-3 هستند. اینفلاماسامِ NLRP-1 عمدتاً در نورونها وجود دارد (26-24). محققان نشان دادند که بیان NLRP-1 در CNS موشهای دیابتی افزایش مییابد و سبب التهاب عصبی ناشی از مقادیر گلوکز بالا میشود (27). گزارش شده است که استرسهای اکسایشی و همچنین بیماریهای تخریب عصبی نظیر آلزایمر فعالیت NLRP-1 را افزایش داده و سبب بروز التهاب نورونی و تخریب اکسونی میشود (28). فعالیت بدنی فواید سلامتی متعددی نظیر افزایش طول عمر، محافظت در برابر بیماریهای قلبی عروقی، دیابت، سرطان و بیماریهای تخریب عصبی دارد (29). محققان نشان دادند که ورزش سبب توسعه یادگیری و حافظه، تأخیر زوال شناختی مرتبط با سن و کاهش خطر تخریب عصب میشود (30). همچنین تحقیقات نشان داده است که فعالیت بدنی علاوه بر توسعه عملکرد رفتاری، سبب ارتقاء شکلپذیری سیناپسی در هیپوکمپ، که یک ساختار کلیدی برای یادگیری است، میشود (31). علاوه بر این، فعالیت بدنی میتواند سطوح سایتوکاینهای پیش التهابی را در مغز کاهش دهد (32). بنابراین، کاهش التهاب مرکزی و محیطی توسط فعالیت بدنی میتواند بهعنوان مکانیزمی متداول برای کاهش خطر هم دیابت و هم زوال شناختی به خدمت گرفته شود (30). در پژوهشهای انجام شده در زمینه فعالیت بدنی و ورزش، گزارش شده است که انجام تمرین استقامتی میتواند دارای اثرات ضداکسایشی نیز باشد و از طریق افزایش میزان آنزیمهای ضداکسایشی، بیماران DM را در مقابل فشار اکسایشی محافظت کند (33). تحقیقات اخیر نشان داده که آسیب اکسایشی در هیپوکمپ هنگام فعالیت با شدت زیر بیشینه کاهش یافته و ورزش منظم میتواند با اثر ضداکسایشی عملکرد حافظه را ارتقاء دهد (35, 34). هرچند ارتباط بیماری DM با اختلالات سیستم عصبی مرکزی و اثرات تخریبی آن بر بافتهای عصبی تا حدودی گزارششده است، با اینحال سازوکار درگیر آن بهخوبی مشخص نیست (37, 36). با توجه به اهمیت اختلالات تخریب عصب در اعمال طبیعی مغز و همچنین اختلالات گزارششده در عملکرد هیپوکمپ بیماران DM، در پژوهش حاضر سعی بر آن است که ارتباط اثر ضدالتهابی و ضداکسایشی فعالیت ورزشی به صورت استقامتی با تغییرات سطوح Panx-1 و پروتئین NLRP-1 بررسی شود. همچنین مطالعه حاضر بهدنبال پاسخگوئی به این سوال است که آیا تمرینات استقامتی قادر است بهعنوان یک راهبرد غیرداروئی این تغییرات احتمالی را تعدیل کند؟ از این رو در مطالعه حاضر، به بررسی تاثیر تمرین استقامتی بر میزان پروتئینهای Panx-1 و NLRP-1 بافت هیپوکمپ موشهای صحرائی نر ویستار دارای دیابت پرداختیم.
روش بررسی
پژوهش حاضر از نوع تجربی و بهشیوه آزمایشگاهی و با طرح پس آزمون آزمون بههمراه گروه کنترل است. جهت انجام آزمایش،32 سر موش صحرایی نر ویستار با 10 هفته سن و میانگین توده بدنی9/4±245 گرم بهعنوان نمونه تحقیق از مرکز نگهداری حیوانات دانشگاه علوم پزشکی لرستان خریداری شد. پس از آشنایی با محیط آزمایشگاه و نوار گردان، 18 سر از رتها از طریق تزریق درون صفاقی STZ (Streptozotocin) دیابتی شده و پس از تأیید القای دیابت و متحمل شدن تلفاتی به تعداد 4 سر رت در 24 ساعت پس از تزریق، 28 رت باقیمانده به روش تصادفی به چهار گروه بدین شرح تقسیم شدند: 1)گروه دیابتی تمرین (DT): این گروه شامل 7 سر رت دیابتی شده بودو از هفته دوازدهم زندگی به مدت6 هفته و هر هفته 5 جلسه تمرین استقامتی انجام میدادند و پس از آخرین جلسه تمرینی تشریح شدند. 2) گروه دیابتی کنترل (DC): این گروه شامل 7 سر رت دیابتی شده بود و در هیچگونه برنامه تمرینی شرکت داده نشدند. این رتها همزمان با بقیه گروهها تشریح شده و کلیه مراحل و آزمایشها مطابق دیگر گروهها بر روی آنها انجام پذیرفت. 3) گروه تمرینی سالم (HT): این گروه شامل 7رت بود که همانند گروه DT در برنامه تمرینی نوارگردان شرکت داده شدند. 4) گروه کنترل سالم (HC): این گروه شامل 7 رت بود که درگیر هیچ فعالیتی نبودند. این رتها (گروه 3 و 4) نیز همزمان با بقیه گروهها تشریح شده و کلیه مراحل و آزمایشها مطابق دیگر گروهها بر روی آنها انجام گرفت. کلیه رتها در شرایط کنترل شده محیطی با میانگین دمای 3±22 درجه سانتیگراد، چرخه روشنایی-تاریکی 12:12 ساعت (شروع چرخه بیداری ساعت 16) و با دسترسی آزاد به آب و غذای ویژه موش نگهداری گردیدند. حیوانات به مدت 2 هفته با شرایط آزمایشگاه و نوار گردان مخصوص جوندگان آشنا شدند. در طول مرحله آشنا سازی، بهمنظور آشنا شدن با شرایط آزمایشگاه، نوار گردان و دستکاری، حیوانات 5 روز در هفته به مدت 15-10 دقیقه با سرعت 10 متر در دقیقه بر روی نوار گردان راه رفتند. تمام جلسات تمرینی در پایان سیکل خواب حیوانات و بین ساعت های 16 تا 18 عصر برگزار گردید. لازم به ذکر است که بهمنظور انجام آزمایشات مولکولی تعداد 5 نمونه از هر گروه مورد سنجش قرار گرفت. موشها در اطاقی به ابعاد 2 متر در 7 متر در شرایط کنترل شده نور (12 ساعت روشنایی و 12 ساعت تاریکی)، دما (3±22 درجه سانتیگراد)، و رطوبت (حدود 45 درصد) نگهداری شدند. تعداد 4 تا 6 عدد موش در قفسهایی از جنس پلکسی گلاس با درب توری نگهداری شدند که آزادانه به آب و غذای استاندارد دسترسی داشتند. پس از 12 ساعت محرومیت از غذا، با تزریق درون صفاقی mg/kg 45محلولSTZ ((Sigma, St. Louis MO, USA، تهیه شده در بافر سیترات تازه 0/5 مولار با pH=4/5) دیابت القاء گردید. بهصورتی که به ازای هر کیلوگرم موش ml2 بافر سیترات به 45 میلیگرم STZ اضافه می شد و با تناسب بستن بین وزن نهایی موش و مقدار بافر سیترات و STZ، حجم نهایی محلول STZ به منظور تزریق مشخص میشد. به رتهای غیر دیابتی نیز معادل حجمی بافر سیترات تزریق گردید. 48 ساعت پس از تزریق، با ایجاد یک جراحت کوچک توسط لانست روی ورید دم رتها، یک قطره خون روی نوار گلوکومتر قرار داده شد و قند خون با استفاده از دستگاه گلوکومتری (Roche Diagnostics K.K., Tokyo, Japan) قرائت گردید. رتهایی که قند خون آنها بالاتر از 300 mg/dl بود، به عنوان رتهای دیابتی در مطالعه حاضر مورد استفاده قرار گرفتند (38). در پژوهش حاضر جهت انجام یک دوره فعالیت استقامتی از پروتکل مطالعه چاء و همکاران (39) استفاده شد؛ بدینصورت که گروههای ورزشی در معرض تمرین نوار گردان برای 5 جلسه در هفته و به مدت 6 هفته قرار گرفتند. سرعت و مدت تمرین نوار گردان بهتدریج افزایش یافت و از 10 متر در دقیقه برای 10 دقیقه در هفته اول، 10 متر در دقیقه برای 20 دقیقه در هفته دوم، 15 متر در دقیقه برای 20 دقیقه در هفته سوم، 15 متر در دقیقه برای 30 دقیقه در هفته چهارم، به 18-17 متر در دقیقه برای 30 دقیقه در هفته پنجم افزایش یافت. جهت رسیدن به سازگاری های بهدست آمده درحالت یکنواخت، تمامی متغیرهای تمرینی درهفته پایانی (هفته ششم) ثابت نگهداشته شدند (جدول1). در پایان 6 هفته برنامه تمرینی، 24 ساعت پس از آخرین جلسه تمرین، رتها توسط تزریق درون صفاقی ترکیب کتامین (mg/kg-175) و زایلازین (mg/kg-1 5) بیهوش شده و پس از جدا کردن سر توسط گیوتین و تحت شرایط استریل بافت هیپوکمپ جدا شده و بلافاصله به فریزر 70- (88FD-2-93-A، ساخت ایران) منتقل شد. ارزیابی بیان پروتئین های Pannexin-1و NLRP-1با استفاده از روش وسترن بلات صورت گرفت. به این صورت که نمونههای بافت هیپوکمپ ابتدا در لایزیس بافر هموژن شدند (لایزیس بافر شامل: Tris-Hcl (0/3 گرم، 50 میلیمول در لیتر) ، تریتونX-100 (0/02گرم، 0/1٪)،کلسیم دی اکسید سدیم (0/05 گرم،0/25٪)،NaCl (0/43 گرم، 150 میلیمول در لیتر)، SDS (0/02 گرم،0/1٪)، اسید اتیلن دی آمین تراستیک (EDTA ، 5/84 گرم) که در 20 میلیلیتر آب مقطر و در7/4=pH مخلوط شده و هموژن شدند (16000 rpm و بهمدت 20 دقیقه در 4 درجهسانتیگراد). برای هر 10 میلیلیتر (10X) از یک قرص مهارکننده پروتئاز استفاده شد. غلظت سوپرنیتان بهدست آمده توسط کیت بردفورد مورد بررسی قرار گرفت. پروتئینها با استفاده از الکتروفورز بر روی ژل SDS-PAGE (10٪) بارگذاری شدند (ابتدا در ولتاژ 60 به مدت 15 دقیقه و سپس در ولتاژ 100 به مدت یک ساعت نگهداری شد). سپس پروتئینها طی فرآیند انتقال بهمدت 105 دقیقه روی کاغذ نیتروسلولوز انتقال داده شدند و سپس با آنتیبادیهای اولیه مونوکلونال Pannexin-1(Human/Mouse/Rat Pannexin-1 Antibody (MAB7097):R&D system.)، NLRP-1(Rat, Human Anti-NALP1 antibody (ab3683): abcam. 100ug) و β-actin (Anti-beta Actin antibody [mabcam 8226]) مورد آزمایش قرار گرفتند (رقت 1: 2000 در PBS). از محلول 5 درصد اسکیم میلک برای مسدود کردن غشا استفاده شد (یک شب) و از آنتیبادی ثانویه (AS09 618 Goat anti-Rat IgG (H&L), HRP conjugated, Agrisera Sweden) نیز برای مدت یک ساعت برای اتصال به آنتیبادی اولیه استفاده شد. باندهای پروتئین در دستگاه Bio-Rad Gel Doc با استفاده از کیت ECL ظاهر شدند. سپس نوارهای پروتئینی با استفاده از نرمافزار Image J در مرحله آخر روش وسترن بلات مورد بررسی قرار گرفت.
تجزیه و تحلیل آماری
ابتدا نرمال بودن توزیع دادهها با استفاده از آزمون شاپیروویلک ارزیابی شد و مشخص شد که توزیع دادهها طبیعی میباشند. در ادامه از آزمون لون (Levene’s Test) برای تعیین همگن بودن واریانسها استفاده شد. برای مقایسه گروهها در متغیرهای مورد مطالعه از تحلیل واریانس یکطرفه استفاده شد. از آزمون توکی بهعنوان آزمون تعقیبی استفاده شد. سطح معناداری نیز P≤ 0/05 در نظر گرفته شد. کلیه بررسیهای آماری با استفاده از نرم افزار version 18 SPSS انجام گرفت.
ملاحظات اخلاقی
پروپوزال این تحقیق توسط کمیته اخلاق دانشگاه لرستان با کد کمیته اخلاق پژوهش بر حیوانات به شماره LU.ECRA.2017.2 مورد بررسی و تایید قرار گرفت.
نتایج
تغییرات وزن بدن و گلوکز خون موشها در گروه¬های مختلف آزمایشی در طی دوره تمرین در شکل 1 و 2 نشان داده شده است. همان¬گونه که در شکل 1 نشان داده شده است وزن اولیه گروهها اختلاف معناداری با یکدیگر نداشت (P>0/05)، اما در پایان پژوهش، میانگین تغییرات وزن موشهای گروه کنترل دیابت نسبت به کنترل سالم و نیز گروه دیابتی تمرین کرده نسبت به گروه کنترل سالم به صورت معناداری کمتر بود (به ترتیب P=0/004 و P=0/003). میانگین تغییرات وزن گروه دیابت تمرین کرده نسبت به گروه سالم تمرین کرده بهصورت معناداری کمتر بود (P=0/003). نتایج تحلیلهای آماری نشان داد که میانگین تغییرات وزن گروههای تمرین کرده سالم نسبت به کنترل سالم معنادار نبود (P=0/09). همچنین، میانگین تغییرات وزن گروههای دیابت تمرین کرده نسبت به کنترل دیابت اگرچه پس از شش هفته تمرین افزایش یافت، اما این افزایش معنادار نبود (P>0/05). همانگونه که در شکل 2 نشان داده شده است در شروع برنامه تمرینی سطح گلوکز خون بهصورت معناداری 48 ساعت پس از القای دیابت توسط استرپتوزوتوسین در موشهای گروه های دیابتی افزایش یافت (P<0/001)، و پس از 6 هفته تمرین استقامتی در مقایسه با گروههای سالم همچنان از اختلاف معنیداری برخوردار بود (P<0/001)، همچنین، در پایان برنامه تمرینی، غلظت گلوکز خون گروه دیابت تمرین کرده نسبت به گروه دیابت کنترل به صورت معناداری پایینتر بود (P<0/001). تغییرات سطوح بیان پروتئینهای Pannexin-1 و NLRP-1، پس از انتهای دوره تمرینی در جدول 2 نشان داده شده است.
شکل3 میزان بیان پروتئین Pannexin-1 را نسبت به β-actin در گروههای مختلف نشان میدهد.
در شکل 4 نمونههایی از روند تغییرات بیان پروتئین Panx-1 در بافت هیپوکمپ گروههای مختلف، که با روش وسترن بلات بر روی کاغذ نیتروسلولز مشخص (Detect) شدهاند، نشان داده شده است. در این تحقیق از سطوح بیان پروتئین β-actin بهعنوان کالیبراتور استفاده شده و تغییرات بیان پروتئینهای مورد نظر با توجه به بیان ثابت این پروتئین ارزیابی گردید. برای کمیسازی باندهای پروتئینی، دانسیته آنها توسط نرمافزار Image J محاسبه شد. نتایج نشان دادکه بیماری دیابت موجب افزایش معنادار سطوح بیان پروتئین Panx-1 در مقایسه با موشهای سالم شد (P<0/05). همچنین یک دوره تمرین استقامتی سطوح بیان این پروتئین را در موشهای دیابتی کاهش داده است (P<0/05). بهعلاوه تمرین استقامتی در موشهای دیابتی سطوح بیان پروتئین Panx-1 تا حدی کاهش داده است که با سطوح این پروتئین در گروه کنترل سالم اختلاف معناداری ندارد (P>0/05).
شکل 5 میزان بیان پروتئین NLRP-1 را نسبت به β-actin در گروه های مختلف نشان میدهد. نتایج بیانگر این نکته است که بیماری دیابت موجب افزایش معنادار سطوح بیان پروتئین NLRP-1 در مقایسه با موش های سالم است (P<0/05). نتایج همچنین نشان داد که یک دوره تمرین استقامتی سطوح این پروتئین را در موشهای دیابتی کاهش داده است (P<0/05). در شکل 6 نمونه هایی از روند تغییرات بیان پروتئین NLRP-1 در بافت هیپوکمپ گروههای مختلف، که با روش وسترن بلات بر روی کاغذ نیتروسلولز مشخص (Detect) شدهاند، نشان داده شده است.
جدول1: نمایش عددی پروتکل در هفتههای مختلف.
جدول 2: میانگین و انحراف معیار تغییرات سطوح بیان پروتئینهای Panx-1 و NLRP-1 در موشهای گروههای مختلف
شکل 1: تغییرات میانگین وزن بدن در موش های گروههای مختلف.
حروف نامتشابه بیانگر وجود اختلاف آماری معنیدار در بین گروهها میباشد (P<0/05).
شکل 2: تغییرات میانگین سطح گلوکز خون در موشهای گروههای مختلف.
حروف نامتشابه بیانگر وجود اختلاف آماری معنیدار در بین گروهها میباشد (P<0/05).
شکل 3: میزان بیان پروتئین Pannexin-1در موش های گروه های مختلف.
حروف نامتشابه بیانگر وجود اختلاف آماری معنیدار در بین گروهها میباشد (P<0/05).
شکل 4 :باندهای وسترنبلات پروتئین Pannexin-1 در بافت هیپوکمپ موشهای گروههای مختلف.
به ترتیب از چپ به راست: DC) گروه دیابت کنترل؛ CO) گروه کنترل سالم؛ TD) گروه دیابت تمرین؛ TH) گروه تمرین سالم.
شکل 5: میزان بیان پروتئین NLRP-1 در موشهای گروههای مختلف.
حروف نامتشابه بیانگر وجود اختلاف آماری معنیدار در بین گروهها میباشد (P<0/05).
شکل 6: مقایسه سطوح بیان پروتئین NLRP-1در بافت هیپوکمپ موشهای گروههای مختلف.
به ترتیب از چپ به راست: DC) گروه دیابت کنترل؛ CO) گروه کنترل سالم؛ TD) گروه دیابت تمرین؛ TH) گروه تمرین سالم
بحث
پژوهش حاضر با هدف بررسی تأثیر یک دوره فعالیت استقامتی بر سطوح بیان پروتئینهای Panx-1 و NLRP-1 در بافت هیپوکمپ موش¬های صحرایینر ویستار دارای دیابت انجام شد. نتایج حاصل از سنجش منظم مقادیر گلوکز خون در طی این تحقیق نشان داد که یک دوره فعالیت بدنی کاهش پایداری را در سطوح گلوکز خون در موشهای صحرایی دیابتی ایجاد میکند. انجمن دیابت آمریکا در سال 2006 به بررسی اثر فعالیت بدنی بر سطح گلوکز پلاسما افراد دیابتی پرداخت. آنها گزارش کردند که ورزش هوازی طولانیمدت بهصورت پایداری سطح گلوکز پلاسما را در افراد مبتلا به دیابت نوع 1 کاهش میدهد (40)، همچنین گوئلفی و همکارانش (2005) نیز در مطالعه خود بیان کردند که هم فعالیت تناوبی با شدت بالا و هم فعالیت با شدت متوسط سطوح گلوکز خون را، در حین تمرین و در زمان بازگشت به حالت اولیه، در افراد مبتلا به دیابت نوع 1 کاهش میدهد (41). فعالیت بدنی پاسخهای هورمون رشد و کاتکولامینها را تحریک کرده و از سقوط گلوکز در حین فعالیت جلوگیری میکند. از سوی دیگر، این امر نشان دهنده این واقعیت است که فعالیت بدنی در زمانی که افزایش تولید گلوکز درون زاد مورد نیاز جبران نمیشود، سبب افزایش بهره برداری از گلوکز میشود (40). نتایج تحقیق حاضر نشان داد که بیماری دیابت منجر به افزایش معنادار سطوح بیان پروتئین Panx-1 در بافت هیپوکمپ موشهای صحرایی نر میشود. نتایج تحقیقات فانگ منگ در سال 2013 نیز نشان داد که بیان Panx-1تحت شرایط هایپرگلیسمیک افزایش مییابد. بنابراین، از آنجا که افزایش بیان این پروتئین سبب راه اندازی آبشارهای سیگنالینگ تولید شاخصهای التهابی میشود، مهار Panx-1 از رهایش فاکتورهای التهابی و آسیبنورونی ناشی از هایپرگلیسمی جلوگیری میکند. از سویی دیگر، علاوه بر نقش ROS (Reactive Oxygen Species)، بهویژه نیتریک اکساید (NO)، در افزایش بازشدن همیکانال Panx-1، ROS در غلظتهای بالا بهعنوان مولکولهای سمی منجر به آسیب بافتی و سپس تغییر یا تخریب انواع سلولها میشود (27). غلظت بالای گلوکز خون در دراز مدت سبب آسیب بافتی می¬شود. اگر گلوکز خون در دیابت مدت زیادی کنترل نشود، عروق خونی بافت¬های متعدد بدن دچار اختلال شده و دستخوش تغییرات ساختمانی می¬شوند که موجب عدم خونرسانی کافی به بافت¬ها می¬گردد. بدین ترتیب خطر حملات قلبی، سکته مغزی، بیماری مرحله نهایی کلیه، رتینوپاتی و کوری، ایسکمی و گانگرن اندام¬ها افزایش می¬یابد (42, 2, 1). این مسئله بسیار مهم است که شرایط ایسکمیک همیکانالهای Panx-1 را در نورونها باز میکند، در واقع تحقیقات نشان داده شده است که رادیکالهای آزاد تولید شده ناشی از شرایط ایسکمیک، از جمله NO، در باز شدن همیکانال Panx-1 نورونی درگیر هستند (43). سایتوکین ها و فاکتورهای رشد فرآیندهای فیزیولوژیکی و پاتولوژیکی متعددی را در سیستم عصبی مرکزی تنظیم میکنند (44). تعدادی از مطالعات نشان میدهند که همیکانالها همچنین توسط سایتوکینها و فاکتورهای رشد تنظیم میشوند. عوامل پیش التهابی مانند TNF-α، IL-1βو پپتید آمیلوئید β منجر به رهایش ATP از طریق همیکانالهای Pannexin و Connexin در آستروسیتها و میکروگلیا میشود (46, 45) و سبب انتشار موج کلسیم به واسطه فعالیت گیرندههای P2 در سلولهای گلیال میشود. از دیگر نتایج این پژوهش می توان به افزایش معنادار سطوح بیان پروتئین NLRP-1 در بافت هیپوکمپ موشهای صحرایینر مبتلا به بیماری دیابت اشاره کرد. این یافته با نتایج فانگ منگ و همکارانش در سال 2014 همسو است. آنها در تحقیق خود بیان کردند که بیان اینفلاماسامِ NLRP-1 در ناحیه کورتکس مغز تحت شرایط هایپرگلیسمیک ناشی از بیماری دیابت تنظیم مثبت شده و به آسیب نورونی منجر شده است (27). فعالیت اینفلاماسامها به عنوان یک ماشین مولکولی درون سلولی برای شروع آسیب نورونی عمل کرده و سرانجام منجر به اختلال عملکرد نورون و آپوپتوزیس می شود (27). این نتایج نشان میدهد که فعالیت اینفلاماسامِ NLRP-1 ممکن است مکانیسم اصلی آغاز آسیب نورونی در شرایط هایپرگلیسمیک باشد. فانگ منگ و همکارانش در تحقیق خود بیان کردند که همیکانال Panx-1 فعالیت اینفلاماسامِ NLRP-1 را در نورون میانجیگری میکند. در واقع، مهار همیکانال Panx-1، التهاب نورونی را در حین شرایط هایپرگلیسمی تضعیف میکند. این یافتهها نشان داد که اینفلاماسامِ NLRP-1 مکانیسم مهمی برای راه اندازی پاسخهای التهابی موضعی در نورونها است (27). نتایج حاصل از تحقیق حاضر نشان داد که یک دوره فعالیت استقامتی منجر به تعدیل سطوح بیان پروتئین های Panx-1 و NLRP-1 و نزدیک ساختن آنها به سطح نرمال، در بافت هیپوکمپ موشهای گروه دیابت تمرین گردید. ورزش و فعالیت بدنی بهعنوان کاهش دهنده پاسخ التهابی سیستمیک مزمن شناخته شده است و اثرات آنتی اکسیدانی و نیز اثرات مثبتی بر شکل پذیری سیناپسی جوندگان دیابتی و یا چاق نشان داده است (47, 30). اسپیک و همکارانش در سال 2014 بهبررسی اثر ضداکسایشی فعالیت ورزشی بر هیپوکمپ پرداختند. آنها نشان داده که آسیب اکسایشی در هیپوکمپ هنگام فعالیت با شدت زیر بیشینه کاهش یافته و ورزش منظم میتواند با اثر ضداکسایشی عملکرد حافظه را ارتقاء دهد (34). بالدوسی و همکارانش به بررسی اثر ضد التهابی فعالیت ورزشی بر روی افراد دیابتی پرداخته و نتایج پژوهش آنها نشان داد که فعالیت بدنی منظم میتواند عوامل التهابی نظیر TNF-α را در جریان گردش خون این بیماران کاهش دهد، همچنیننشان داد که فعالیت بدنی سبب کاهش ROS و افزایش سطوح دفاع ضداکسایشی میشود (48). همچنین چیریکو و همکارانش در سال 2016 کاهش معنیداری را در IL-1β (سایتوکین پردازش شده توسط اینفلاماسامها) در مغز موشهای تیمار شده با رژیم پرچرب، پس از وادار کردن حیوان به تمرین استقامتی، نشان دادند (49). در هیپوکمپ موشهای دیابتی شده با STZ، نه تنها افزایش بارز گونههای اکسیژن فعال مشاهده شده است، بلکه همچنین فعالیت پایدار NF-KB قابل مشاهده است (51, 50). فعال شدن NF-KBمیتواند منجر به تولید محصولات سایتوکسیک شود که التهاب و استرس اکسیداتیو را تشدید کرده و باعث اختلال در عملکرد سلول و متعاقب آن افزایش آپوپتوزیس یا مرگ سلولی میشود (52). از سویی بهخوبی نشان داده شده است که در حین فرآیندهای التهابی در بیماریهای تخریب عصب نظیر دیابت، باز شدن همیکانالها ایمنی سلولهای عصبی را کاهش میدهد (44). همانگونه که در بیماریهایی نظیر دیابت میلیتوس مشاهده میشود فرآیندهای تخریب عصب، که با التهاب عصبی همراه هستند، ممکن است باعث افزایش فعالیت همیکانالهای آستروگلیال و نورونی شود، که منجر به مرگ سلول و تخریب عملکرد دستگاه عصبی مرکزی میشود (53). از سوی دیگر در وضعیت پاتولوژیکی نظیر شرایط ایسکمیک، جایی که رادیکالهای آزاد به ویژه NO افزایش مییابند، همیکانالهای غشایی Panx-1 در حالت باز شده قرار دارند (44) و آبشار سیگنالینگی را راه اندازی میکنند که نهایتاً منجر به مرگ سلولی میشود (53). علاوه بر این که باز شدن همیکانالهای غشایی خود عامل تحریکی برای فعال شدن اینفلاماسامها است، اتصال ATP ترشح شده از سلولهای التهابی به کانالهای P2X، جریان یونی Na+، Ca2+ و K+را در عرض منافذ به راه میاندازد (ورود بیشتر Ca2+ فعالیت آبشارهای نوروتوکسیک درون سلولی را افزایش میدهد) که کمپلکس اینفلاماسام-کاسپاز-1 را فعال کرده و سبب شکسته شدن سایتوکینهای پیشالتهابی به اشکال فعال آنها مانند IL-1β و IL-18 میشود، که قادر هستند نوعی از مرگ سلولی برنامهریزی شده که با نام پریوپتوزیس (خودکشی سلولی) شناخته شده است را میانجیگری کنند (54). بنابراین، سازگاری ناشی از فعالیت بدنی با شدت متوسط و یا با شدت زیر بیشینه احتمالاً میتواند با کاهش رهایش شاخصهای التهابی و نیز کاهش رادیکالهای آزاد و آسیبهای اکسایشی، فعالیت همیکانالهای غشایی مانند Panx-1 و متعاقب آن فعالیت اینفلاماسامهایی نظیر NLRP-1 را مهار کند. باید خاطر نشان کرد که علاوه بر این که عواملی نظیر شاخصهای التهابی بیان این دو پروتئین را افزایش میدهند، افزایش بیان این دو پروتئین بهصورت چرخهای معیوب، خود سبب افزایش شاخصهای التهابی و تسریع فرآیند مرگ سلولی میشوند (55). علاوه بر فواید قلبی عروقی فعالیت بدنی و همپوشانی آنها با سلامت مغز، اثرات فعالیت بدنی بر جلوگیری از ناهنجاریهای تخریب عصب و گسترش بی رویه دارو درمانی نیز میتواند جالب توجه باشد (56). نشان داده شده است که Panx-1 در پاسخ به محرومیت از اکسیژن و گلوکز (شرایط ایسکمیک) در نورونهای هیپوکمپ یک جریان ثانویۀ شدید را بهوجود میآورد که غشاء را دپولاریزه میکند (دپلاریزاسیون اکسیژن)، این شرایط در نهایت منجر به مرگ نورونی میشود (22). افزایش شبکه عروقی و جریان خون ناشی از فعالیت بدنی در بیماران دیابتی ممکن است سبب در دسترس قرار گرفتن بیشتر اکسیژن در بافت مغزی شود. در نتیجه، احتمالاً در دسترس قرار داشتن اکسیژن سبب مهار بیان بیش از اندازه این همیکانال Panx-1شده و از راهاندازی چرخه معیوب بیان کمپلکس پروتئینی NLRP-1 و شاخصهای التهابی در سلولهای آستروگلیال و نورونهای بافت هیپوکمپ موشهای صحرایی دیابتی جلوگیری میکند. در این تحقیق تنها بیان این پروتئینها در بافت هیپوکمپ مورد ارزیابی قرار گرفته است، مطالعات بیشتری نیاز است تا عوامل گوناگون موثر در بیان چنین پروتئینهایی را در اجزای مختلف سیستم عصبی مرکزی مورد بررسی قرار دهد.
نتیجهگیری
نتایج این پژوهش نشان داد که فعالیت استقامتی با شدت متوسط و یا زیر بیشینه میتواند مهار کننده خوبی برای کنترل بیان بیش از اندازه پروتئینهای Panx-1 و NLRP-1 در بافت هیپوکمپ موشهای دیابتی باشد. چنین مهاری میتواند از راهاندازی چرخههای معیوب درون سلولی جلوگیری کرده و به بیان دیگر شرایط تحریک کننده تخریب عصبی را کنترل کند.
سپاسگزاری
این پژوهش حاصل رساله دکتری رشته فیزیولوژی ورزشی (گرایش عصبی- عضلانی) دانشگاه لرستان است که در تاریخ 27/11/1395 و با شماره 968/تگ به تصویب رسیده است. بدین وسیله از معاونت پژوهشی دانشگاه لرستان و همکاری دانشگاه شهید چمران اهواز تشکر و قدردانی میشود. هزینه اجرای این پژوهش از محل اعتبارات پژوهانه واحد پژوهشی دانشگاه لرستان تامین شده است.
حامی مالی: معاونت محترم پژوهشی دانشگاه لرستان.
تعارض در منافع: وجود ندارد.
References:
1-Goossens MM, Nelson RW, Feldman EC, Griffey SM. Response to Insulin Treatment and Survival in 104 Cats with Diabetes Mellitus (1985–1995). J Vet Intern Med 1998; 12(1): 1-6.
2-Rand JS, Fleeman LM, Farrow HA, Appleton DJ, Lederer R. Canine and Feline Diabetes Mellitus: Nature or Nurture? J Nutr 2004; 134(8 Suppl): 2072S-2080S.
3-Maritim AC, Sanders RA, Watkins JB. Diabetes, Oxidative Stress, And Antioxidants: A Review. J Biochem Mol Toxiol 2003; 17(1): 24-38.
4-Sima AA, Zhang W, Xu G, Sugimoto K, Guberski D, Yorek MA. A Comparison of Diabetic Polyneuropathy in Type II Diabetic BBZDR/Wor Rats and in Type I Diabetic BB/Wor Rats. Diabetologia 2000; 43(6): 786-93.
5-Li ZG, Zhang W, Grunberger G, Sima AA. Hippocampal Neuronal Apoptosis in Type 1 Diabetes. Brain Res 2002; 946(2): 221-31.
6-Martínez-Tellez R, Gómez-Villalobos MJ, Flores G. Alteration in Dendritic Morphology of Cortical Neurons in Rats with Diabetes Mellitus induced by Streptozotocin. Brain Res 2005; 1048(1-2): 108-15.
7-Reagan LP. Insulin Signaling Effects on Memory and Mood. Curr Opin Pharmacol 2007; 7(6): 633-7.
8-Reagan LP, Magariños AM, McEwen BS. Neurological Changes induced by Stress in Streptozotocin Diabetic Rats. Ann N Y AcadSci 1999; 893(1): 126-37.
9-den Heijer T, Vermeer S, van Dijk E, Prins N, Koudstaal PJ, Hofman A, et al. Type 2 Diabetes and Atrophy of Medial Temporal Lobe Structures on Brain MRI. Diabetologia 2003; 46(12): 1604-10.
10-Sima AA, Nathaniel V, Bril V, McEwen TA, Greene DA. Histopathological Heterogeneity of Neuropathy in Insulin-Dependent and Non-Insulin-Dependent Diabetes, And Demonstration of Axo-Glial Dysjunction in Human Diabetic Neuropathy. J Clin Invest 1988; 81(2): 349-364.
11-Toth C. Diabetes and Neurodegeneration in the Brain. Handb Clin Neurol 2013; 126: 489-511.
12-Baydas G, Reiter RJ, Yasar A, Tuzcu M, Akdemir I, Nedzvetskii VS. Melatonin Reduces Glial Reactivity in the Hippocampus, Cortex, And Cerebellum of Streptozotocin-Induced Diabetic Rats. Free Radic Biol Med 2003; 35(7): 797-804.
13-Kalalian-Moghaddam H, Baluchnejadmojarad T, Roghani M, Goshadrou F, Ronaghi A.Hippocampal Synaptic Plasticity Restoration and Anti-Apoptotic Effect Underlie Berberine Improvement of Learning and Memory in Streptozotocin-Diabetic Rats. Eur j pharmacol 2013; 698(1): 259-66.
14-Zhang X, Xu L, He D, Ling Sh. Endoplasmic Reticulum Stress-Mediated Hippocampal Neuron Apoptosis involved in Diabetic Cognitive Impairment. Biomed Res Int 2013; 2013: 924327.
1-Goossens MM, Nelson RW, Feldman EC, Griffey SM. Response to Insulin Treatment and Survival in 104 Cats with Diabetes Mellitus (1985–1995). J Vet Intern Med 1998; 12(1): 1-6.
2-Rand JS, Fleeman LM, Farrow HA, Appleton DJ, Lederer R. Canine and Feline Diabetes Mellitus: Nature or Nurture? J Nutr 2004; 134(8 Suppl): 2072S-2080S.
3-Maritim AC, Sanders RA, Watkins JB. Diabetes, Oxidative Stress, And Antioxidants: A Review. J Biochem Mol Toxiol 2003; 17(1): 24-38.
4-Sima AA, Zhang W, Xu G, Sugimoto K, Guberski D, Yorek MA. A Comparison of Diabetic Polyneuropathy in Type II Diabetic BBZDR/Wor Rats and in Type I Diabetic BB/Wor Rats. Diabetologia 2000; 43(6): 786-93.
5-Li ZG, Zhang W, Grunberger G, Sima AA. Hippocampal Neuronal Apoptosis in Type 1 Diabetes. Brain Res 2002; 946(2): 221-31.
6-Martínez-Tellez R, Gómez-Villalobos MJ, Flores G. Alteration in Dendritic Morphology of Cortical Neurons in Rats with Diabetes Mellitus induced by Streptozotocin. Brain Res 2005; 1048(1-2): 108-15.
7-Reagan LP. Insulin Signaling Effects on Memory and Mood. Curr Opin Pharmacol 2007; 7(6): 633-7.
8-Reagan LP, Magariños AM, McEwen BS. Neurological Changes induced by Stress in Streptozotocin Diabetic Rats. Ann N Y AcadSci 1999; 893(1): 126-37.
9-den Heijer T, Vermeer S, van Dijk E, Prins N, Koudstaal PJ, Hofman A, et al. Type 2 Diabetes and Atrophy of Medial Temporal Lobe Structures on Brain MRI. Diabetologia 2003; 46(12): 1604-10.
10-Sima AA, Nathaniel V, Bril V, McEwen TA, Greene DA. Histopathological Heterogeneity of Neuropathy in Insulin-Dependent and Non-Insulin-Dependent Diabetes, And Demonstration of Axo-Glial Dysjunction in Human Diabetic Neuropathy. J Clin Invest 1988; 81(2): 349-364.
11-Toth C. Diabetes and Neurodegeneration in the Brain. Handb Clin Neurol 2013; 126: 489-511.
12-Baydas G, Reiter RJ, Yasar A, Tuzcu M, Akdemir I, Nedzvetskii VS. Melatonin Reduces Glial Reactivity in the Hippocampus, Cortex, And Cerebellum of Streptozotocin-Induced Diabetic Rats. Free Radic Biol Med 2003; 35(7): 797-804.
13-Kalalian-Moghaddam H, Baluchnejadmojarad T, Roghani M, Goshadrou F, Ronaghi A.Hippocampal Synaptic Plasticity Restoration and Anti-Apoptotic Effect Underlie Berberine Improvement of Learning and Memory in Streptozotocin-Diabetic Rats. Eur j pharmacol 2013; 698(1): 259-66.
14-Zhang X, Xu L, He D, Ling Sh. Endoplasmic Reticulum Stress-Mediated Hippocampal Neuron Apoptosis involved in Diabetic Cognitive Impairment. Biomed Res Int 2013; 2013: 924327.
15-Ristow M. Neurodegenerative Disorders Associated with Diabetes Mellitus. J Mol Med (Berl) 2004; 82(8): 510-29.
16-Bond SR, Wang N, Leybaert L, Naus CC. Pannexin 1 Ohnologs in the Teleost Lineage. J Membr Biol 2012; 245(8): 483-93.
17-Wang H, Cao Y, Chiang CY, Dostrovsky JO, Sessle BJ. The Gap Junction Blocker Carbenoxolone Attenuates Nociceptive Behavior and Medullary Dorsal Horn Central Sensitization induced by Partial Infraorbital Nerve Transection in Rats. Pain 2014; 155(2): 429-35.
18-Penuela S, Gehi R, Laird DW. The Biochemistry and Function of Pannexin Channels. Biochim Biophys Acta 2013; 1828(1): 15-22.
19-Isakson BE, Thompson RJ. Pannexin-1 as a Potentiator of Ligand-Gated Receptor Signaling. Channels (Austin) 2014; 8(2): 118-23.
16-Bond SR, Wang N, Leybaert L, Naus CC. Pannexin 1 Ohnologs in the Teleost Lineage. J Membr Biol 2012; 245(8): 483-93.
17-Wang H, Cao Y, Chiang CY, Dostrovsky JO, Sessle BJ. The Gap Junction Blocker Carbenoxolone Attenuates Nociceptive Behavior and Medullary Dorsal Horn Central Sensitization induced by Partial Infraorbital Nerve Transection in Rats. Pain 2014; 155(2): 429-35.
18-Penuela S, Gehi R, Laird DW. The Biochemistry and Function of Pannexin Channels. Biochim Biophys Acta 2013; 1828(1): 15-22.
19-Isakson BE, Thompson RJ. Pannexin-1 as a Potentiator of Ligand-Gated Receptor Signaling. Channels (Austin) 2014; 8(2): 118-23.
20-Pasquier F, Boulogne A, Leys D, Fontaine P. Diabetes Mellitus and Dementia. Diabetes Metab 2006; 32(5 Pt 1): 403-14.
21-LaFerla FM, Green KN, Oddo S. Intracellular Amyloid-Beta in Alzheimer's Disease. Nat rev Neurosci 2007; 8(7): 499-509.
22-Thompson RJ, Zhou N, MacVicar BA. Ischemia Opens Neuronal Gap Junction Hemichannels. Science 2006; 312(5775): 924-7.
23-Martinon F, Burns K, Tschopp J. The Inflammasome: A Molecular Platform Triggering Activation of Inflammatory Caspases and Processing of Proil-Β. Mol Cell 2002; 10(2): 417-26.
24-de Rivero Vaccari JP, Dietrich WD, Keane RW. Activation and Regulation of Cellular Inflammasomes: Gaps in our Knowledge for Central Nervous System Injury. J Cereb Blood Flow Metabol 2014; 34(3): 369-75.
25-Kummer JA, Broekhuizen R, Everett H, Agostini L, Kuijk L, Martinon F, et al. Inflammasome Components NALP 1 and 3 Show Distinct but Separate Expression Profiles in Human Tissues Suggesting a Site-Specific Role in the Inflammatory Response. J Histochem Cytochem 2007; 55(5): 443-52.
26-Salminen A, Ojala J, Suuronen T, Kaarniranta K, Kauppinen A. Amyloid‐Β Oligomers Set Fire to Inflammasomes and Induce Alzheimer's Pathology. J Cell Mol Med 2008; 12(6a): 2255-62.
27-Meng XF, Wang XL, Tian XJ, Yang ZH, Chu GP, Zhang J, et al. Nod-Like Receptor Protein 1 Inflammasome Mediates Neuron Injury under High Glucose. Mol Neurobiol 2014; 49(2): 673-84.
28-Kaushal V, Dye R, Pakavathkumar P, Foveau B, Flores J, Hyman B, et al. Neuronal NLRP1 Inflammasome Activation of Caspase-1 Coordinately Regulates Inflammatory Interleukin-1-Beta Production and Axonal Degeneration-Associated Caspase-6 Activation. Cell Death Differ 2015; 22(10): 1676-86.
29-Handschin C, Spiegelman BM. The Role of Exercise and PGC1α in Inflammation and Chronic Disease. Nature 2008; 454(7203): 463-69.
30-Cotman CW, Berchtold NC. Physical Activity and the Maintenance of Cognition: Learning from Animal Models. Alzheimers Dement 2007; 3(2 Suppl): S30-7.
31-Farmer J, Zhao X, Van Praag H, Wodtke K, Gage FH, Christie BR. Effects of Voluntary Exercise on Synaptic Plasticity and Gene Expression in the Dentate Gyrus of Adult Male Sprague–Dawley Rats in Vivo. Neuroscience 2004; 124(1): 71-9.
32-Weisman D, Hakimian E, Ho GJ. Interleukins, Inflammation, And Mechanisms of Alzheimer's Disease.Vitam Horm 2006; 74: 505-30.
33-Coskun O, Ocakci A, Bayraktaroglu T, Kanter M. Exercise Training Prevents and Protects Streptozotocin-Induced Oxidative Stress and Βeta-Cell Damage in Rat Pancreas. Tohoku J Exp Med 2004; 203(3): 145-54.
34-Speck AE, Tromm CB, Pozzi BG, Paganini CS, Tuon T, Silveira PC, et al. The Dose-Dependent Antioxidant Effects of Physical Exercise in the Hippocampus of Mice.Neurochem Res 2014; 39(8): 1496-501.
35-Bertram S, Brixius K, Brinkmann C. Exercise for the Diabetic Brain: How Physical Training may Help Prevent Dementia and Alzheimer’s Disease in T2DM Patients.Endocrine 2016; 53(2): 350-63.
36-Meek TH, Morton GJ. Leptin, Diabetes, And the Brain. Indian J Endocrinol Metab 2012; 16(Suppl 3): S534-S542.
37-Ristow M. Neurodegenerative Disorders Associated with Diabetes Mellitus. J Mol Med (Berl) 2004; 82(8): 510–29.
38-Szkudelski T. The Mechanism of Alloxan and Streptozotocin Action in B Cells of the Rat Pancreas. Physiol Res 2001; 50(6): 537-46.
39-Chae CH, Jung SL, An SH, Jung CK, Nam SN, Kim HT. Treadmill Exercise Suppresses Muscle Cell Apoptosis by Increasing Nerve Growth Factor Levels and Stimulating P-Phosphatidylinositol 3-Kinase Activation in the Soleus of Diabetic Rats. J Physiol Biochem 2011; 67(2): 235-41.
40-Taplin CE, Cobry E, Messer L, McFann K, Chase HP, Fiallo-Scharer R. Preventing Post-Exercise Nocturnal Hypoglycemia in Children with Type 1 Diabetes. J Pediatr 2010; 157(5): 784-8.
41-Guelfi KJ, Jones TW, Fournier PA. The Decline in Blood Glucose Levels is less with Intermittent High-Intensity Compared with Moderate Exercise in Individuals with Type 1 Diabetes. Diabetes Care 2005; 28(6): 1289-94.
42-Hall JE. Guyton and Hall Textbook of Medical Physiology. 13th ed. Elsevier Health Sciences; 2015; 78-96.
43-Zhang L, Deng T, Sun Y, Liu K, Yang Y, Zheng X. Role for Nitric Oxide in Permeability of Hippocampal Neuronal Hemichannels during Oxygen Glucose Deprivation. J Neurosci Res 2008; 86(10): 2281-91.
44-Orellana JA, Martinez AD, Retamal MA. Gap Junction Channels and Hemichannels in the CNS: Regulation by Signaling Molecules. Neuropharmacology 2013; 75: 567-82.
45-Bennett MV, Garré JM, Orellana JA, Bukauskas FF, Nedergaard M, Giaume C, et al. Connexin and Pannexin Hemichannels in Inflammatory Responses of Glia and Neurons. Brain Res 2012; 1487: 3-15.
46-Orellana JA, Shoji KF, Abudara V, Ezan P, Amigou E, Sáez PJ, et al. Amyloid Β-Induced Death in Neurons Involves Glial and Neuronal Hemichannels. J Neurosci 2011; 31(13): 4962-77.
47-Churchill JD, Galvez R, Colcombe S, Swain RA, Kramer AF, Greenough WT. Exercise, Experience and the Aging Brain. Neurobiol Aging 2002; 23(5): 941-55.
48-Balducci S, Zanuso S, Nicolucci A, Fernando F, Cavallo S, Cardelli P, et al.Anti-Inflammatory Effect of Exercise Training in Subjects with Type 2 Diabetes and the Metabolic Syndrome is Dependent on Exercise Modalities and Independent of Weight Loss. Nutr Metab Cardiovasc Dis 2010; 20(8): 608-17.
49-Cai M, Wang H, Li JJ, Zhang YL, Xin L, Li F, et al. The Signaling Mechanisms of Hippocampal Endoplasmic Reticulum Stress Affecting Neuronal Plasticity-Related Protein Levels in High Fat Diet-Induced Obese Rats and the Regulation of Aerobic Exercise. Brain Behav Immun 2016; 57: 347-59.
50- Alvarez-Nölting R, Arnal E, Barcia JM, Miranda M, Romero FJ. Protection by DHA of Early Hippocampal Changes in Diabetes: Possible Role of CREB and NF-ΚB. Neurochem Res 2012; 37(1): 105-15.
51-Muriach M, Bosch-Morell F, Alexander G, Blomhoff R, Barcia J, Arnal E, et al. Lutein Effect on Retina and Hippocampus of Diabetic Mice. Free Radic Biol Med 2006; 41(6): 979-84.
52-Morgan MJ, Liu Z-g. Crosstalk of Reactive Oxygen Species and NF-ΚB Signaling. Cell Research 2011; 21(1): 103-15.
53-Orellana JA, Froger N, Ezan P, Jiang JX, Bennett MV, Naus CC, et al. ATP and Glutamate Released via Astroglial Connexin 43 Hemichannels Mediate Neuronal Deaththrough Activation of Pannexin 1 Hemichannels. J Neurochem 2011; 118(5): 826-40.
54- Schroder K, Tschopp J. The Inflammasomes. Cell 2010; 140(6): 821-32.
55-Bernier LP. Purinergic Regulation of Inflammasome Activation after Central Nervous System Injury. J Gen Physiol 2012; 140(5): 571-5.
56-Wang S, Chen L, Zhang L, Huang C, Xiu Y, Wang F, et al. Effects of Long-Term Exercise on Spatial Learning, Memory Ability, And Cortical Capillaries in Aged Rats. Med Sci Monit 2015; 21: 945-54.
21-LaFerla FM, Green KN, Oddo S. Intracellular Amyloid-Beta in Alzheimer's Disease. Nat rev Neurosci 2007; 8(7): 499-509.
22-Thompson RJ, Zhou N, MacVicar BA. Ischemia Opens Neuronal Gap Junction Hemichannels. Science 2006; 312(5775): 924-7.
23-Martinon F, Burns K, Tschopp J. The Inflammasome: A Molecular Platform Triggering Activation of Inflammatory Caspases and Processing of Proil-Β. Mol Cell 2002; 10(2): 417-26.
24-de Rivero Vaccari JP, Dietrich WD, Keane RW. Activation and Regulation of Cellular Inflammasomes: Gaps in our Knowledge for Central Nervous System Injury. J Cereb Blood Flow Metabol 2014; 34(3): 369-75.
25-Kummer JA, Broekhuizen R, Everett H, Agostini L, Kuijk L, Martinon F, et al. Inflammasome Components NALP 1 and 3 Show Distinct but Separate Expression Profiles in Human Tissues Suggesting a Site-Specific Role in the Inflammatory Response. J Histochem Cytochem 2007; 55(5): 443-52.
26-Salminen A, Ojala J, Suuronen T, Kaarniranta K, Kauppinen A. Amyloid‐Β Oligomers Set Fire to Inflammasomes and Induce Alzheimer's Pathology. J Cell Mol Med 2008; 12(6a): 2255-62.
27-Meng XF, Wang XL, Tian XJ, Yang ZH, Chu GP, Zhang J, et al. Nod-Like Receptor Protein 1 Inflammasome Mediates Neuron Injury under High Glucose. Mol Neurobiol 2014; 49(2): 673-84.
28-Kaushal V, Dye R, Pakavathkumar P, Foveau B, Flores J, Hyman B, et al. Neuronal NLRP1 Inflammasome Activation of Caspase-1 Coordinately Regulates Inflammatory Interleukin-1-Beta Production and Axonal Degeneration-Associated Caspase-6 Activation. Cell Death Differ 2015; 22(10): 1676-86.
29-Handschin C, Spiegelman BM. The Role of Exercise and PGC1α in Inflammation and Chronic Disease. Nature 2008; 454(7203): 463-69.
30-Cotman CW, Berchtold NC. Physical Activity and the Maintenance of Cognition: Learning from Animal Models. Alzheimers Dement 2007; 3(2 Suppl): S30-7.
31-Farmer J, Zhao X, Van Praag H, Wodtke K, Gage FH, Christie BR. Effects of Voluntary Exercise on Synaptic Plasticity and Gene Expression in the Dentate Gyrus of Adult Male Sprague–Dawley Rats in Vivo. Neuroscience 2004; 124(1): 71-9.
32-Weisman D, Hakimian E, Ho GJ. Interleukins, Inflammation, And Mechanisms of Alzheimer's Disease.Vitam Horm 2006; 74: 505-30.
33-Coskun O, Ocakci A, Bayraktaroglu T, Kanter M. Exercise Training Prevents and Protects Streptozotocin-Induced Oxidative Stress and Βeta-Cell Damage in Rat Pancreas. Tohoku J Exp Med 2004; 203(3): 145-54.
34-Speck AE, Tromm CB, Pozzi BG, Paganini CS, Tuon T, Silveira PC, et al. The Dose-Dependent Antioxidant Effects of Physical Exercise in the Hippocampus of Mice.Neurochem Res 2014; 39(8): 1496-501.
35-Bertram S, Brixius K, Brinkmann C. Exercise for the Diabetic Brain: How Physical Training may Help Prevent Dementia and Alzheimer’s Disease in T2DM Patients.Endocrine 2016; 53(2): 350-63.
36-Meek TH, Morton GJ. Leptin, Diabetes, And the Brain. Indian J Endocrinol Metab 2012; 16(Suppl 3): S534-S542.
37-Ristow M. Neurodegenerative Disorders Associated with Diabetes Mellitus. J Mol Med (Berl) 2004; 82(8): 510–29.
38-Szkudelski T. The Mechanism of Alloxan and Streptozotocin Action in B Cells of the Rat Pancreas. Physiol Res 2001; 50(6): 537-46.
39-Chae CH, Jung SL, An SH, Jung CK, Nam SN, Kim HT. Treadmill Exercise Suppresses Muscle Cell Apoptosis by Increasing Nerve Growth Factor Levels and Stimulating P-Phosphatidylinositol 3-Kinase Activation in the Soleus of Diabetic Rats. J Physiol Biochem 2011; 67(2): 235-41.
40-Taplin CE, Cobry E, Messer L, McFann K, Chase HP, Fiallo-Scharer R. Preventing Post-Exercise Nocturnal Hypoglycemia in Children with Type 1 Diabetes. J Pediatr 2010; 157(5): 784-8.
41-Guelfi KJ, Jones TW, Fournier PA. The Decline in Blood Glucose Levels is less with Intermittent High-Intensity Compared with Moderate Exercise in Individuals with Type 1 Diabetes. Diabetes Care 2005; 28(6): 1289-94.
42-Hall JE. Guyton and Hall Textbook of Medical Physiology. 13th ed. Elsevier Health Sciences; 2015; 78-96.
43-Zhang L, Deng T, Sun Y, Liu K, Yang Y, Zheng X. Role for Nitric Oxide in Permeability of Hippocampal Neuronal Hemichannels during Oxygen Glucose Deprivation. J Neurosci Res 2008; 86(10): 2281-91.
44-Orellana JA, Martinez AD, Retamal MA. Gap Junction Channels and Hemichannels in the CNS: Regulation by Signaling Molecules. Neuropharmacology 2013; 75: 567-82.
45-Bennett MV, Garré JM, Orellana JA, Bukauskas FF, Nedergaard M, Giaume C, et al. Connexin and Pannexin Hemichannels in Inflammatory Responses of Glia and Neurons. Brain Res 2012; 1487: 3-15.
46-Orellana JA, Shoji KF, Abudara V, Ezan P, Amigou E, Sáez PJ, et al. Amyloid Β-Induced Death in Neurons Involves Glial and Neuronal Hemichannels. J Neurosci 2011; 31(13): 4962-77.
47-Churchill JD, Galvez R, Colcombe S, Swain RA, Kramer AF, Greenough WT. Exercise, Experience and the Aging Brain. Neurobiol Aging 2002; 23(5): 941-55.
48-Balducci S, Zanuso S, Nicolucci A, Fernando F, Cavallo S, Cardelli P, et al.Anti-Inflammatory Effect of Exercise Training in Subjects with Type 2 Diabetes and the Metabolic Syndrome is Dependent on Exercise Modalities and Independent of Weight Loss. Nutr Metab Cardiovasc Dis 2010; 20(8): 608-17.
49-Cai M, Wang H, Li JJ, Zhang YL, Xin L, Li F, et al. The Signaling Mechanisms of Hippocampal Endoplasmic Reticulum Stress Affecting Neuronal Plasticity-Related Protein Levels in High Fat Diet-Induced Obese Rats and the Regulation of Aerobic Exercise. Brain Behav Immun 2016; 57: 347-59.
50- Alvarez-Nölting R, Arnal E, Barcia JM, Miranda M, Romero FJ. Protection by DHA of Early Hippocampal Changes in Diabetes: Possible Role of CREB and NF-ΚB. Neurochem Res 2012; 37(1): 105-15.
51-Muriach M, Bosch-Morell F, Alexander G, Blomhoff R, Barcia J, Arnal E, et al. Lutein Effect on Retina and Hippocampus of Diabetic Mice. Free Radic Biol Med 2006; 41(6): 979-84.
52-Morgan MJ, Liu Z-g. Crosstalk of Reactive Oxygen Species and NF-ΚB Signaling. Cell Research 2011; 21(1): 103-15.
53-Orellana JA, Froger N, Ezan P, Jiang JX, Bennett MV, Naus CC, et al. ATP and Glutamate Released via Astroglial Connexin 43 Hemichannels Mediate Neuronal Deaththrough Activation of Pannexin 1 Hemichannels. J Neurochem 2011; 118(5): 826-40.
54- Schroder K, Tschopp J. The Inflammasomes. Cell 2010; 140(6): 821-32.
55-Bernier LP. Purinergic Regulation of Inflammasome Activation after Central Nervous System Injury. J Gen Physiol 2012; 140(5): 571-5.
56-Wang S, Chen L, Zhang L, Huang C, Xiu Y, Wang F, et al. Effects of Long-Term Exercise on Spatial Learning, Memory Ability, And Cortical Capillaries in Aged Rats. Med Sci Monit 2015; 21: 945-54.
نوع مطالعه: پژوهشي |
موضوع مقاله:
فیزیولوژی ورزش
دریافت: 1398/9/2 | پذیرش: 1399/4/9 | انتشار: 1399/4/9
دریافت: 1398/9/2 | پذیرش: 1399/4/9 | انتشار: 1399/4/9
ارسال پیام به نویسنده مسئول
| بازنشر اطلاعات | |
 |
این مقاله تحت شرایط Creative Commons Attribution-NonCommercial 4.0 International License قابل بازنشر است. |
