

دوره 31، شماره 11 - ( بهمن 1402 )
جلد 31 شماره 11 صفحات 7193-7179 |
برگشت به فهرست نسخه ها


![]()
![]()
![]()
Download citation:
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:



BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
Khalili A. A Review of Primary Immuno-deficiency Disorders Predisposing to Cancer. JSSU 2024; 31 (11) :7179-7193
URL: http://jssu.ssu.ac.ir/article-1-6006-fa.html
URL: http://jssu.ssu.ac.ir/article-1-6006-fa.html
خلیلی عباس. مروری بر نقایص ایمنی اولیه مستعد به کانسر. مجله علمي پژوهشي دانشگاه علوم پزشكي شهید صدوقی يزد. 1402; 31 (11) :7179-7193



متن کامل [PDF 1011 kb]
(560 دریافت)
| چکیده (HTML) (1669 مشاهده)
References:
1- Khalili A. A Review of Primary Immunodeficiency Diseases with Skin Manifestations. J Shahid
Sadoughi Uni Med Sci 2022; 29(10): 4164-79.
2- Salavoura K, Kolialexi A, Tsangaris G, Mavrou A. Development of Cancer in Patients with Primary Immunodeficiencies. Anticancer Res 2008; 28(2B): 1263-9.
3- Rezaei N, Hedayat M, Aghamohammadi A, Nichols KE. Primary Immunodeficiency Diseases Associated with Increased Susceptibility to Viral Infections and Malignancies. J Allergy Clin Immunol 2011; 127(6): 1329-41.
4- Kebudi R, Kiykim A, Sahin MK. Primary Immunodeficiency and Cancer in Children; A Review of the Literature. Curr Pediatr Rev 2019; 15(4): 245-50.
5- Mayor PC, Eng KH, Singel KL, Abrams SI, Odunsi K, Moysich KB, et al. Cancer in Primary Immunodeficiency Diseases: Cancer Incidence in the United States Immune Deficiency Network Registry. J Allergy Clin Immunol 2018; 141(3): 1028-35.
6- Matza Porges S, Shamriz O. Genetics of Immune Dysregulation and Cancer Predisposition: Two Sides of the Same Coin. Clin Exp Immunol 2022; 210(2): 114-27.
7- Yazdani R, Habibi S, Sharifi L, Azizi G, Abolhassani H, Olbrich P، et al. Common Variable Immunodeficiency: Epidemiology، Pathogenesis، Clinical Manifestations، Diagnosis، Classification، and Management. J Investig Allergol Clin Immunol 2020; 30(1): 14-34.
8- Bruns L, Panagiota V, von Hardenberg S, Schmidt G, Adriawan IR, Sogka E, et al. Common Variable Immunodeficiency-Associated Cancers: The Role of Clinical Phenotypes، Immunological and Genetic Factors. Front Immunol 2022; 13: 742530.
9- Gullo I, Costa C, Silva SL, Ferreira C, Motta A, Silva SP, et al. The Dysfunctional Immune System in Common Variable Immunodeficiency Increases the Susceptibility to Gastric Cancer. Cells 2020; 9(6): 1498.
10- Kiaee F, Azizi G, Rafiemanesh H, Zainaldain H, Sadaat Rizvi F, Alizadeh M, et al. Malignancy in Common Variable Immunodeficiency: A Systematic Review and Meta-Analysis. Expert Rev Clin Immunol 2019; 15(10): 1105-13.
11- Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin 2018; 68(6): 394-424.
12- Leone P, Vacca A, Dammacco F, Racanelli V. Common Variable Immunodeficiency and Gastric Malignancies. Int J Mol Sci 2018; 19(2): 451.
13- Pulvirenti F, Pecoraro A, Cinetto F, Milito C, Valente M, Santangeli E, et al. Gastric Cancer is the Leading Cause of Death in Italian Adult Patients with Common Variable Immunodeficiency. Front Immunol 2018; 9: 2546.
14- Tak Manesh A, Azizi G, Heydari A, Kiaee F, Shaghaghi M, Hossein-Khannazer N, et al. Epidemiology and Pathophysiology of Malignancy in Common Variable Immunodeficiency? Allergol Immunopathol (Madr) 2017; 45(6): 602-15.
15- Khalili A, Yadegari AH, Delavari S, Yazdani R, Abolhassani H. Disseminated Intravascular Coagulation Associated with Large Deletion of Immunoglobulin Heavy Chain. Iran J Allergy Asthma Immunol 2021; 20(6): 778-83.
16- Khalili A, Plebani A, Vitali M, Abolhassani H, Lougaris V, Mirminachi B, et al. Autosomal Recessive Agammaglobulinemia: A Novel Non-Sense Mutation in CD79a. J Clin Immunol 2014; 34(2): 138-41.
17- Bagheri Y, Vosughi A, Azizi G, Yazdani R, Kiaee F, Hafezi N, et al. Comparison of Clinical and Immunological Features and Mortality in Common Variable Immunodeficiency and Agammaglobulinemia Patients. Immunol Lett 2019; 210: 55-62.
18- Hajjar J, Hasan S, Forbes LR, Hemmige V, Orange JS. Gastric Adenocarcinoma in a Patient with X-Linked Agammaglobulinemia and HIV: Case Report and Review of the Literature. Front Pediatr 2016; 4: 100.
19- Brosens LA, Tytgat KM, Morsink FH, Sinke RJ, Ten Berge IJ, Giardiello FM, et al. Multiple Colorectal Neoplasms in X-Linked Agammaglobulinemia. Clinical Gastroenterology and Hepatology 2008; 6(1): 115-9.
20- Gokce G, Ceylan OM, Uysal Y, Yildizoglu U, Atas E, Kurt B. Epiphora as the Presenting Sign of Relapsed Non-Hodgkin Lymphoma in a Child with Bruton Agammaglobulinemia. Eur J Ophthalmol 2015; 25(1): 65-7.
21- Moeini Shad T, Yazdani R, Amirifar P, Delavari S, Heidarzadeh Arani M, Mahdaviani SA, et al. Atypical Ataxia Presentation in Variant Ataxia Telangiectasia: Iranian Case-Series and Review of the Literature. Front Immunol 2022; 12: 779502.
22- Mitiagin Y, Barzilai A. Ataxia-Telangiectasia Mutated Plays an Important Role in Cerebellar Integrity and Functionality. Neural Regen Res 2023; 18(3): 497-502.
23- Bakhtiar S, Salzmann-Manrique E, Donath H, Woelke S, Duecker RP, Fritzemeyer S, et al. The Incidence and Type of Cancer in Patients with Ataxia‐Telangiectasia Via a Retrospective Single‐Centre Study. Br J Haematol 2021; 194(5): 879-87.
24- Oska S, Zarbo A, Yeager D, Friedman BJ, Shwayder T. Melanoma Arising in a Patient with Ataxia-Telangiectasia: A Call for Full Skin Examinations in This Patient Population. Pediatr Dermatol 2020; 37(4): 767-8.
25- Hosahalli Vasanna S, Pereda MA, Dalal J. Clinical Features، Cancer Biology، Transplant Approach and other Integrated Management Strategies for Wiskott–Aldrich Syndrome. J Multidiscip Healthc 2021; 14: 3497-512.
26- Sullivan KE, Mullen CA, Blaese RM, Winkelstein JA. A Multiinstitutional Survey of the Wiskott-Aldrich Syndrome. J Pediatr 1994; 125(6 Pt 1): 876-85.
27- Menotti M, Ambrogio C, Cheong TC, Pighi C, Mota I, Cassel SH, et al. Wiskott–Aldrich Syndrome Protein (WASP) is a Tumor Suppressor in T Cell Lymphoma. Nat Med 2019; 25(1): 130-40.
28- Perry GS 3rd, Spector BD, Schuman LM, Mandel JS, Anderson VE, McHugh RB, et al. The Wiskott-Aldrich Syndrome in the United States and Canada (1892–1979). J Pediatr 1980; 97(1): 72-8.
29- Cekic S, Metin A, Aytekin C, Edeer Karaca N, Baris S, Karali Y, et al. The Evaluation of Malignancies in Turkish Primary Immunodeficiency Patients; A Multicenter Study. Pediatr Allergy Immunol 2020; 31(5): 528-36.
30- Tsilifis C, Freeman AF, Gennery AR. STAT3 Hyper-Ige Syndrome—An Update and Unanswered Questions. J Clin Immunol 2021; 41(5): 864-80.
31- Haskologlu S, Kostel Bal S, Islamoglu C, Aytekin C, Guner S, Sevinc S, et al. Clinical، Immunological Features and Follow Up of 20 Patients with Dedicator of Cytokinesis 8 (DOCK8) Deficiency. Pediatr Allergy Immunol 2020; 31(5): 515-27.
32- Aydin SE, Kilic SS, Aytekin C, Kumar A, Porras O, Kainulainen L, et al. DOCK8 Deficiency: Clinical and Immunological Phenotype and Treatment Options-A Review of 136 Patients. J Clin Immunol 2015; 35(2): 189-98.
33- Sharapova SO, Pashchenko OE, Bondarenko AV, Vakhlyarskaya SS, Prokofjeva T, Fedorova AS, et al. Geographical Distribution، Incidence، Malignancies، and Outcome of 136 Eastern Slavic Patients with Nijmegen Breakage Syndrome and NBN Founder Variant C. 657_661del5. Front Immunol 2021; 11: 602482.
34- El Hasbaoui B، Elyajouri A، Abilkassem R، Agadr A. Nijmegen Breakage Syndrome: Case Report and Review of Literature. Pan Afr Med J 2020; 35: 85.
35- Sharma R, Lewis S, Wlodarski MW. DNA Repair Syndromes and Cancer: Insights into Genetics and Phenotype Patterns. Front Pediatr 2020; 8: 570084.
36- Izawa K, Martin E, Soudais C, Bruneau J, Boutboul D, Rodriguez R, et al. Inherited CD70 Deficiency in Humans Reveals a Critical Role for the CD70–CD27 Pathway in Immunity to Epstein-Barr Virus Infection. J Exp Med 2017; 214(1): 73-89.
37- Abolhassani H, Edwards ES, Ikinciogullari A, Jing H, Borte S, Buggert M, et al. Combined Immunodeficiency and Epstein-Barr Virus–Induced b Cell Malignancy in Humans with Inherited CD70 Deficiency. J Exp Med 2017; 214(1): 91-106.
38- Abolhassani H. Specific Immune Response and Cytokine Production in CD70 Deficiency. Frontiers in Pediatrics 2021; 9: 615724.
39- Han BK, Olsen NJ, Bottaro A. The CD27–CD70 Pathway and Pathogenesis of Autoimmune Disease. Semin Arthritis Rheum 2016: 45(4): 496-501.
40- Ghosh S, Köstel Bal S, Edwards ESJ, Pillay B, Jiménez Heredia R, Erol Cipe F, et al. Extended Clinical and Immunological Phenotype and Transplant Outcome in CD27 and CD70 Deficiency. Blood 2020; 136(23): 2638-55.
41- Lino CNR, Ghosh S. Epstein–Barr Virus in Inborn Immunodeficiency—More than Infection. Cancers (Basel) 2021; 13(19): 4752.
42- Xu T, Zhao Q, Li W, Chen X, Xue X, Chen Z, et al. X-Linked Lymphoproliferative Syndrome in Mainland China: Review of Clinical، Genetic، and Immunological Characteristic. Eur J Pediatr 2020; 179(2): 327-38.
43- El-Mallawany NK, Curry CV, Allen CE. Haemophagocytic Lymphohistiocytosis and Epstein–Barr Virus: A Complex Relationship with Diverse Origins، Expression and Outcomes. Br J Haematol 2022; 196(1): 31-44.
44- Szmyd B, Mlynarski W, Pastorczak A. Genetic Predisposition to Lymphomas: Overview of Rare Syndromes and Inherited Familial Variants. Mutat Res Rev Mutat Res 2021; 788: 108386.
45- Liang JH, Zhu HY, Xu DM, Wang L, Wang Y, Qiao C, et al. A New SH2D1A Mutation in a Female Adult XLP Disease with Hemophagocytic Lymphohistiocytosis and NK-Cell Leukemia. Ann Hematol 2019; 98(12): 2829-31.
46- Abbas R, Larisch S. Targeting XIAP for Promoting Cancer Cell Death—The Story of ARTS and SMAC. Cells 2020; 9(3): 663.
47- Mudde ACA, Booth C, Marsh RA. Evolution of Our Understanding of XIAP Deficiency. Front Pediatr 2021; 9: 660520.
48- Latour S, Winter S. Inherited Immunodeficiencies with High Predisposition to Epstein–Barr Virus-Driven Lymphoproliferative Diseases. Front Immunol 2018; 9: 1103.
49- Duan L, Grunebaum E. Hematological Malignancies Associated with Primary Immunodeficiency Disorders. Clinical Immunology 2018; 194: 46-59.
50- Wallace JG, Alosaimi MF, Khayat CD, Jaber F, Almutairi A, Beaussant-Cohen S, et al. ITK Deficiency Presenting as Autoimmune Lymphoproliferative Syndrome. J Allergy Clin Immunol 2021; 147(2): 743-5. e1.
51- Ghosh S, Bienemann K, Boztug K, Borkhardt A. Interleukin-2-Inducible T-Cell Kinase (ITK) Deficiency-Clinical and Molecular Aspects. J Clin Immunol 2014; 34(8): 892-9.
52- Consonni F, Gambineri E, Favre C. ALPS، FAS، and Beyond: From Inborn Errors of Immunity to Acquired Immunodeficiencies. Ann Hematol 2022; 101(3): 469-84.
53- Li P, Huang P, Yang Y, Hao M, Peng H, Li F. Updated Understanding of Autoimmune Lymphoproliferative Syndrome (ALPS). Clin Rev Allergy Immunol 2016; 50(1): 55-63.
54- Hafezi N, Zaki-Dizaji M, Nirouei M, Asadi G, Sharifinejad N, Jamee M, et al. Clinical، Immunological، and Genetic Features in 780 Patients with Autoimmune Lymphoproliferative Syndrome (ALPS) and ALPS‐Like Diseases: A Systematic Review. Pediatr Allergy Immunol 2021; 32(7): 1519-32.
55- Leechawengwongs E, Shearer WT. Lymphoma Complicating Primary Immunodeficiency Syndromes. Curr Opin Hematol 2012; 19(4): 305-12.
56- Heusinkveld LE, Majumdar S, Gao JL, McDermott DH, Murphy PM. WHIM Syndrome: From Pathogenesis Towards Personalized Medicine and Cure. Journal of Clinical Immunology 2019; 39(6): 532-56.
57- Geier CB, Ellison M, Cruz R, Pawar S, Leiss-Piller A, Zmajkovicova K, et al. Disease Progression of WHIM Syndrome in an International Cohort of 66 Pediatric and Adult Patients. J Clin Immunol 2022; 42(8): 1748-65.
58- Tiri A, Masetti R, Conti F, Tignanelli A, Turrini E, Bertolini P, et al. Inborn Errors of Immunity and Cancer. Biology (Basel) 2021; 10(4): 313.
59- Lazzareschi I, Rossi E, Curatola A, Capozio G, Benacquista L, Iezzi L, et al. Assessment of Congenital Neutropenia in Children: Common Clinical Sceneries and Clues for Management. Mediterr J Hematol Infect Dis 2022; 14(1): e2022008.
60- Hojabri M, Farsi Y, Jamee M, Abolhassani H, Khani HHK, Karimi A, et al. JAGN1 Mutation with Distinct Clinical Features; Two Case Reports and Literature Review. BMC Pediatr 2023; 23(1): 206.
61- Link DC. Mechanisms of Leukemic Transformation in Congenital Neutropenia. Curr Opin Hematol 2019; 26(1): 34-40.
متن کامل: (1383 مشاهده)
مقدمه
نقایص ایمنی اولیه اختلالات نسبتاً نادر و هتروژنی هستند که یک یا چندین عضو از سیستم ایمنی دچار اختلال عملکرد میشود. تظاهرات بالینی افراد مبتلا به این بیماریها بسیار متغیر و متنوع هستند. اکثریت این افراد با توجه به نوع اختلالی که دارند دچار عفونتهای مکرر با جرمهای مختلف میشوند. این افراد به دلایل مختلفی ممکن است مستعد بیماریهای بدخیم باشند (1). نوع بدخیمی بستگی دارد به نوع نقص ایمنی اولیه، سن بیمار و نوع ویروسی که بیمار به آن مبتلا میشود. لذا مکانیسمهای پاتوژنیک مختلفی در بروز این بدخیمیها نقش دارند. تصور میشود پاسخ ایمونولوژیک میزبان نسبت به تحریک مزمن آنتیژنیک در بروز کانسر مهمترین نقش را داشته باشد (2). پیشرفتهای بسیار مهمی در خصوص درک مکانیسمهای مولکولی نقایص سیستم ایمنی سلولار و هومورال صورت گرفته است که دانستههای ما را درباره بروز انواع عفونتها و تومورها درهریک از این اختلالات افزایش داده و در تشخیص زودهنگام این عوارض به ما کمک میکند (3). شواهد متعددی وجود دارد که نشان میدهد سیستم ایمنی در بروز بسیاری از بدخیمیها نقش مهمی دارد. از اینرو نقایص ایمنی از دسته اختلالاتی هستند که ممکن است فرد را به کانسر مبتلا کنند. مکانیسمهای متعددی در بروز بدخیمی در بیماران مبتلا به نقص ایمنی اولیه دخالت دارند: ازجمله نقصهایی در آپوپتوزیس و تمایز سلولی مانند اختلالات سیتواسکلتون، متابولیسم سلولی، سیتوتوکسیسیتی. همچنین ناهنجاریهایی در ماده ژنتیکی سلول مثل ناپایداری کروموزومی، و اشکال در ترمیم DNA نیز در ایجاد کانسر دخالت دارند. از سوی دیگر عوامل خارجی مثل عفونت با ویروسهای انکوژن به عنوان مثال (Epstein-Barr virus) EBV و (Human papilloma virus)HPV و همچنین وجود التهاب بافتی مزمن، فاکتورهایی هستند که فرد مبتلا به نقص ایمنی را مستعد بدخیمی خواهند کرد (4). تقریباً در10% سرطانها استعداد خانوادگی مشاهده میشود. این در حالی است که در 80%-60% سرطانهای خانوادگی بررسی ژنتیکی صورت نمیگیرد. در مطالعهای که روی 3658 بیمار مبتلا به نقص ایمنی اولیه بین سالهای 2015-2003 صورت گرفته بود میزان خطر نسبی سرطان در مقایسه با جمعیت عمومی 1/24 گزارش شد. در جنس مذکر میزان این خطر 1/19بود ولی در جنس مونث میزان خطر نسبی در گروه نقایص ایمنی اولیه و جمعیت عمومی یکسان بود (5). در برخی خانوادهها اتوایمیونیتی و استعداد به عفونت دیده میشود. در سالهای اخیر رابطه اثبات شدهای بین دیس رگولیشن سیستم ایمنی و کانسر گزارش شده است. بنابراین جای تعجبی ندارد که بیماران دارای اختلال سیستم ایمنی مستعد به کانسر باشند (6). ما در این مطالعه قصد داریم مروری کوتاه بر جنبههای اپیدمیولوژیک، پاتوفیزیولوژیک و بالینی نقایص سیستم ایمنی اولیه داشته باشیم که انواع مختلف بدخیمی در آنها بوجود میآیند.
روشبررسی
جستجوی مقالات در پایگاه های اطلاعاتی google scholar،pubmed، web of science ،scopus صورت گرفت. در این بررسی سعی شده است از جدیدترین و معتبرترین مقالات مرتبط با موضوع که به زبان انگلیسی به چاپ رسیده است استفاده شود. برای انسجام و نظم بیشتر مطالب سعی شده عناوین مورد بحث براساس تقسیم بندی IUIS 2023 مطرح شود.
Humoral immunedeficiency
(common variable immunodeficiency)(CVID): شایعترین نقص ایمنی هومورال اولیه علامتدار بیماری (CVID) است. این بیماری گروهی از اختلالات هتروژن را در برمیگیرد که در تولید آنتی بادی نقص دارند. شایعترین تظاهر بالینی بیماران مبتلا به CVID عفونتهای سینوپولمونری و گوارشی میباشد. باکتریهایی مثل گونههای استرپتوکک، هموفیلوس آنفولانزا، موراکسلا کاتارالیس، نایسریا و گونههای استافیلوکک میگروارگانیسمهای شایع در این افراد هستند. همچنین پاتوژنهایی مثل مایکوپلاسما، رینوویروسها، سیتومگالوویروس و پنوموسیستیس ژیروویسی اگر چه در این افراد شایع نیست ولی گهگاهی دیده میشوند (7). این بیماران در کنار استعداد به عفونتهای مختلف مستعد یکسری بیماریهای غیر عفونی نیز میباشند. مثل: بیماریهای خود ایمنی، اختلالات اتواینفلیمیتوری و اختلالات لنفوپرولیفراتیو پلی کلونال. میزان پریواالانس بدخیمی در این نوع اختلال حدود 8/6% تخمین زده میشود (8). این بیماران 47- 7 برابر شانس بروز کانسر نسبت به جمعیت عمومی دارند. لنفوم (40/5%) و آدنوکارسینوم معده (7/5%) شایعترین بدخیمیهای گزارش شده در بیماران مبتلا به CVID هستند. سایر بدخیمیها مثل کانسر پستان، پوست، تیموس، لوکمی، و ملانوم در ردههای بعدی قرار دارند. کانسر معده اولین علت مرگ مرتبط با بدخیمی در بین بیماران CVID میباشد. این در حالیست که در جمعیت عمومی پنجمین علت مرگ به حساب میآید (13-9). میزان بروز سرطان در بیماران مبتلا، در دهههای چهارم تا ششم بیشترین مقدار است. مکانیسمهای پاتوفیزیولوژیک ایجاد بدخیمی در بیماران مبتلا به CVID به طور کامل مشخص نشده است. اما تصور میشود مکانیسمهایی چون ناپایداری و استعداد ژنتیکی، فعالیت و تکثیر مداوم و افزایش یافته سلولهای لنفوییدی طی دورههای عفونت و اختلال در کلیرانس ویروسهای اونکوژنیک و عفونتهای باکتریال در ایجاد بدخیمی نقش داشته باشند. سلولهای کلیدی در کنترل سلولهای تومورال، NK cell ها و لنفوسیتهای CD8+ سیتوتوکسیک هستند. همچنین سلولهای CD4+ هم بهخصوص TH1 cellها با کمک به فعالشدن CD8+سلها در کنترل سلولهای بدخیم میتوانند نقش ایفا کنند. اختلال درپاسخهای ایمونولوژیک سلولهای CD4+ وCD8+ و همچنین کاهش سلولهای (Natural killer cell) NK cell در بیماران(Common variable immunodeficiency) CVID میتوانند توجیهکننده بروز بیماریهای بدخیم باشند (14).
(X-linked agammaglobulinemia)(XLA): آگاماگلوبولینمیای وابسته به X برای اولین بار در سال 1952 توسط شخصی بنام بروتون تشخیص داده شد. دراین افراد بهدلیل اختلال در تمایز پیشسازهای لنفوسیت B در مغز استخوان، لنفوسیتهای B خون محیطی و پلاسماسلها به شدت کاهش یافته هستند و به تبع آن تمام ردههای ایمونوگلوبولینها افت شدید دارند. فرمهای اوتوزومال مغلوب بیماری نیز گزارش شده است که حاصل موتاسیون در ژنهای µ heavy chain، Igα، Igβ،λ5 ، BLNK، PIK3R1 میباشند (16،15،7). در مطالعهای که توسط آقامحمدی و همکاران صورت گرفته بود تظاهرات بالینی، یافتههای ایمونولوژیک و عوارض در بیماریXLA و CVID مورد مقایسه قرار گرفتند و مشخص شد همه موارد بدخیمی در XLA نسبت به CVID کمتر میباشد (17). علی رغم اینکه بدخیمیها در بیماران XLA نادر است اما کیس ریپورتهایی در مورد وجود آدنوکارسینوم معده که احتمالاً به علت گاستریت مزمن ناشی از هلیکوباکتر پیلوری میباشد گزارش شده است. سایر بدخیمیهایی که در این افراد مشاهده شده شامل(Acute lymphocytic leukemia ) ALL – لنفوما – آدنوکارسینوم کولورکتال میباشد ( 20-18).
(cellular and combined immunodeficiency)
Ataxia telangiectasia: اتاکسی تلانژکتازی یک بیماری نادر و مولتیسیستمی است با توارث اتوزومال مغلوب که با موتاسیون در ژنataxia telangiectasia mutated gene(ATM) مشخص میشود. محصول این ژن پروتئینکینازی است که در کنترل سیکل سلولی و ترمیم مولکول DNA دخالت دارد. فرم کلاسیک بیماری بهصورت شروع زودرس آتاکسی مخچهای، تلانژکتازی، عفونت مکرر ناشی از نقص ایمنی، رادیوسنسیتیویتی، استعداد به بدخیمی و افزایش آلفافیتوپروتئین تظاهر مییابد. اکثریت بیماران در دهه اول بیماری وابسته به ویلچر میشوند و در دهه دوم زندگی به دلیل نارسایی تنفسی یا بدخیمی فوت خواهند کرد (22،21). در این بیماران ناپایداری کروموزومی ناشی از اختلال در ژن ATM وجود دارد که فرد را مستعد رادیوسنسیتیویتی و بدخیمی میکند. حتی کاریرهای هتروزیگوس ژن ATM نیز به خاطر استعداد به بدخیمی کاهش امید به زندگی دارند. آتاکسی تلانژکتازی به دو فرم تظاهر مییابد. یکی فرم کلاسیک و شدید که تظاهرات بالینی زودرس در دوران کودکی دارند. وارینت دیگری هم وجود دارد که خفیفتر است و تظاهرات بیماری در سنین نوجوانی و بزرگسالی بروز پیدا میکند. در مطالعهای که توسط Emily Petley و همکارانش صورت گرفت 1826 بدخیمی را در 1643 بیمار مبتلا به اتاکسی تلانژکتازی مورد بررسی قرار دادند. شایعترین بدخیمی در گروه کلاسیک، نانهوچکین لنفوما با سن متوسط 9 سال و 8 ماه بود. دومین بدخیمی لوکمی لنفوسیتیک حاد با سن متوسط 11 سال بود و در رده سوم لنفوم هوچکین قرار داشت. در گروه وارینت شایعترین کانسر سرطان پستان و بهدنبال آن لوکمی گزارش شد (23). بر اساس مطالعات مختلف شایعترین بدخیمیها در این افراد بدخیمیهای هماتولوژیک به خصوص فرمهای لنفوییدی است. اما برخی solid tumorها مانند سرطان معده، پستان وپوست (ملانوما) هم در این بیماران گزارش شده است (24).
(Wiscott – Aldrich syndrome)(WSA): سندرم ویسکوت آلدریچ یک بیماری نادر وابسته به x است که در سال 1937 میلادی گزارش شد. بیماران با تریاد میکروترومبوسیتوپنی، عفونت مکرر و اگزما تظاهر پیدا میکنند. این افراد همچنین مستعد خودایمنی و بدخیمیهای لنفوییدی نیز هستند. ژن WAS روی بازوی کوتاه کروموزوم xقرار گرفته و پروتئینی را به نام WASP در سلولهای بنیادین مغز استخوان، رده لنفوییدی و میلوییدی کد میکند. نقش این پروتئین در پلیمریزاسیون اکتین و بازسازی اسکلت سلولی است. همچنین تنظیمکننده سیناپس ایمونولوژیک سلولهای B وT نیز میباشد. از آنجاییکه WASP انحصارا روی سلولهای هماتوپوئتیک اکسپرس میشود، لذا بیماران مبتلا به ویسکوتآلدریچ همراه با اختلالاتی در سیستم ایمنی و هماتوپوئتیک هستند. پروتئین WASP توسطWIP (WASP-interacting-protein) تنظیم میشود. مطالعات نشان داده است لنفوسیتهای T در بیماران مبتلا به ویسکوت آلدریچ و موشهایی که کمبود WASP و WIP دارند درT cell receptor signaling اشکال دارند. شواهد نشان میدهد که این اختلال در پاتوژنز لنفوم T cell نقش مهمی دارد. این بیماران مستعد عفونتهای سینوپولمونری، ویروسی (هرپس و واریسلا)، عفونتهای کاندیدایی و عفونتهای فرصتطلب مثل پنوموسیستیس ژیروویسی هستند. شایعترین بدخیمی در این افراد سرطانهای هماتولنفوییدی است. این بدخیمیها در سنین نوجوانی و بزرگسالی شیوع بیشتری دارند تا در کودکان. بدخیمیهای ناشایعترکه در این افراد گزارش شده است شامل گلیوما، آکوستیک نوروما و کارسینوم بیضه بوده است (27-25). در مطالعهای perry و همکارانش خطر نسبی (Relative risk) کانسر را در بیماران مبتلا 100 برابر جمعیت عمومی گزارش کردند که با افزایش سن، میزان خطر افزایش می یافت (28). در مطالعه دیگری که Sukru Cekic و همکارانش روی 6392 بیمار مبتلا به نقص ایمنی انجام دادند 59 بیمار همراه با بدخیمی را مورد ارزیابی قرار دادند. فرکانس بدخیمی در ویسکوتالدریچ 9/1% گزارش شد (29).
Hyper IgE syndrome: سندرم hyper IgE جزء گروهی از خطاهای ارثی ایمنی است که با استعداد به عفونتهای ریوی و پوستی، اگزما و افزایش شدید سطح سرمی IgE مشخص میشوند. فرم کلاسیک آن ناشی از موتاسیون در ژن کدکننده STAT3 (signal transduction and activator of transcription 3) میباشد. این بیماران مستعد عفونتهای استافیلوککی پوستی و ریوی هستند. عفونتهای کاندیدایی هم در این بیماران به دلیل اختلال در محورIL17-TH17 مشاهده میشود. تظاهرات غیر عفونی این بیماران شامل آلرژی، اختلالات بافت همبند (صورت خشن، پل بینی پهن، تاخیر در افتادن دندانهای شیری، شکنندگی استخوانها، هیپراکستنسی بیلیتی (Hyper extensibility) مفاصل و...)، اختلالات عروقی، خودایمنی و بالاخره استعداد به بدخیمی میباشد. 7% بیماران مبتلا به موتاسیون ژن STAT3 دچار بدخیمی میشوند که شایعترین فرم آن بیماری لنفوم میباشد (30). فرم اتوزومال مغلوب یماری hyper IgE بیماری DOCK8 deficiency است. آتوپی (آلرژی غذایی- درماتیت و آسم)، عفونتهای سینوپولموناری و عفونتهای ویروسی در پوست، بدخیمیهای زودرس، افزایش IgE و ائوزینوفیلی خون محیطی از جمله علائم شایع در این افراد است (31). مطالعهای توسط Susanne E روی 136 بیمار مبتلا به DOCK8 صورت گرفت. میزان بدخیمی در این بیماران 17% گزارش شد. شایعترین بدخیمی انواع هماتولوژیک بود (48%)، کانسرهای اپیتلیال (39%) و سایر بدخیمیها (22%) در ردههای بعدی قرار داشتند (32).
Nijmegen breakage syndrome (NBS): سندرم NBS یک بیماری نادر اتوزومال مغلوب است. ناپایداری کروموزومی مشخصه بارز بیماری به حساب میآید. شیوع بیماری در نواحی اروپای مرکزی و اروپای شرقی بیشتراست (لهستان، جنوب آلمان، جمهوری چک، اوکراین). موارد اسپورادیک هم در خاور میانه گزارش شده است (33). این افراد میکروسفالی بدو تولد همراه با اختلال ساختاری صورت به شکل bird-like و کاهش ضریب هوشی بدون سایر تظاهرات نورولوژیک دارند. سایر علائم آنها شامل استعداد به عفونتهای مکرر، اختلال رشد خفیف، نارسایی زودرس تخمدانها و تمایل شدید به بیماریهای بدخیمی بهخصوص بدخیمیهای خونی میباشد. رادیوسنسیتیویتی و ناپایداری کروموزومی از خصوصیات بارز این بیماران است. وجود موتاسیون در هر دو آلل ژن nibrin (NBN) تشخیص NBS را کامل میکند (34). نقص ایمنی در این بیماران به شکل کاهش سلولهای Tو B(80%)، هیپوگاماگلوبولینمیای شدید (20%) و IgA deficiency (50%) میباشد. بدخیمی علت مهم مرگ و میر در این افراد است. بیش از 40% بیماران تا سن 20 سالگی مبتلا به کانسر میشوند که اغلب منشا لنفوییدی دارد (diffuse large B cell lymphoma،T cell lymphoblastic lymphoma). سایر بدخیمیهای هماتولوژیک در این افراد شامل هوچکین،ALL T & B cell و AML میباشد. تومورهای solid درNBS نیز بهصورت نادر دیده میشوند که عبارتند از مدولوبلاستوما، رابدومیوسارکوما، کارسینوم پاپیلاری تیروئید، گلیوما، مننژیوما، نوروبلاستوما و یوئینگسارکوما (35). تحقیقی توسط Svetlana. O روی 136 بیمار مبتلا به NBS انجام شد. بدخیمی در 45/6% افراد مورد بررسی گزارش شد. نسبت مرد به زن 1/5 به 1 بود. نئوپلاسمهای لنفوییدی اکثریت بدخیمیها را به خود اختصاص داد (9/91%). لنفوم نان هوچکین شایعترین بدخیمی بود که 2/3 موارد را شامل میشد. در حالیکه لنفوم هوچکین با فرکانس 3/3% فرم نادرتری در NBS بود. تومورهای solid فقط 5/6% کل نئوپلاسمها را در بر میگرفت (33).
Diseases of immune dysregulation
CD27-CD70 deficiency: مولکولهای CD70 و CD27 متعلق به خانواده بزرگ رسپتورهای TNFمیباشند و نقش مهمی در ایجاد و حفظ ایمنی سلولی دارند. این مسیر ایمونولوژیک در ایمنی ضد ویروسی نقش مهمی دارد. اخیراً مشخص شده که اختلال در این مولکولها فرد را مستعد عوارض لنفوپرولیفراتیو ویروس EBV میکند. بیماران مبتلا به کمبود CD27 استعداد بسیار زیادی به اختلالات لنفوپرولیفراتیو ناشی از عفونت EBV دارند (37،36). مولکول CD70 یک لیگاند کمک تحریکی (co-stimulatory) است که روی تعدادی از سلولهای ایمونولوژیک مانند T cellها وB cellها و سلولهای دندریتیک بیان میشود. اتصال این مولکول با رسپتورش، CD27، باعث فعال شدن سلولهای ایمونولوژیک CD27+ شده میزان بقا و تکثیر آنها را افزایش میدهد. تعامل بین CD27/CD70 برای تکامل B cellها الزامی نیست اما به نظر میرسد که در فعال شدن B cellها، تولید آنتیبادی و تشکیل ژرمینال سنتر نقش مهمی داشته باشد. یافتههای بالینی و ایمونولوژیک بیمارانی که در محور CD27-CD70 اختلال دارند شامل وجود نقص ایمنی اولیه با دیسرگولیشن سیستم ایمنی، اشکال در تکامل B cellهای ترمینال، اتوایمیونیتی، عفونتهای ویروسی شدید و لنفوما میباشد (39،38،4). شایعترین تظاهرات بالینی بیماران با اختلال محور CD27-CD70 وابسته به عفونت EBV است. در مطالعهای که توسط Sujal Ghosh روی 49 بیمار صورت گرفت 37% بیماران اختلال لنفوپرولیفراتیو، 43% لنفوما و 18% HLH داشتند. تست مثبت EBV در بدو تشخیص، در 31 فرد از 33 بیمار مبتلا به CD27 deficiency و 15 نفر از 16 بیمار مبتلا به CD70 deficency گزارش شد (40). یافتههای بالینی هیپوگاماگلوبولینمیا در بیماران با کمبود CD27 و CD70 ناشی از دیسرگولیشن سیستم ایمنی با واسطه ویروس EBV میباشد. این بیماران میتوانند با مونوکلئوزیس شدید – هموفاگوسیتیک لنفوهیستیوسیتوزیس (HLH) – اختلالات لنفوپرولیفراتیو و لنفوم تظاهر یابند. در مطالعهای 11 بیمار از 21 بیمار مبتلا به CD70- deficiency و 36% (12/33) از بیماران CD27-deficient دچار لنفوم شده بودند و شایعترین فرم آن لنفوم هوچکین بود. مکانیسمهای دقیق این بینظمیهای ایمونولوژیک بهطور کامل مشخص نیست اما به نظر میرسد که CD27 و CD70 در کنترل ایمونولوژیک بدخیمیها جدای از وجود عفونت EBV نقش مهمی دارند (41).
(سندرم دونکان)x-linked lymphoproliferative syndrome(XLP): بیماری XLP یک نقص ایمنی اولیه نادری است که با استعداد زیاد به عفونت EBV – دیس گاماگلوبولینمی – اختلالات لنفوپرولیفراتیو و هموفاگوسیتیک لنفوهیستیوسیتوزیس تظاهر پیدا میکند. این بیماری برای اولین بار در سال 1970 گزارش شد. در حالیکه اولین ژن مسبب بیماری کهSH2D1A میباشد و کد کنندهSLAM- associated protein ((SAP است، در سال 1998 کشف شد. دومین ژن بیماری XIAP در سال 2006 در بیمارانی پیدا شد که علائم شبیه XLPداشتند ولی موتاسیون درژن SH2D1A وجود نداشت. بنابراین XLP به دو دسته تقسیم شد: 1- SAP- deficiency(XLP1) که موتاسیون در ژن SH2D1A وجود دارد. 2-XIAP- deficiency(XLP2) (42). مولکول SAP تنظیمکننده فعالیت B cellها و همچنین تنظیمکننده فعالیت لیتیک T cell ها میباشد. فعالیت پروآپوپتوتیک SAP باعث جلوگیری از تکثیر سلولهای آسیب دیده میشود که این خود در پیشگیری از ایجاد کانسر نقش کلیدی دارد. از طرفی اشکال در بقاء سلولهای خونساز ناشی از اختلال در پاسخهای سیتوتوکسیک T و NKسلها با واسطه رسپتورSLAM، ممکن است منجر به بروز بدخیمی گردد. نقص SAP در سلولهای CD8 منجر به اختلال در چسبندگی و نابودی B cell های فعال شده بهویژه B cellهای تغییر ماهیت یافته توسط EBV میشود که سطوح بالای SLAM را بیان میکنند. اختلال در بقا ایمونولوژیک B cell ممکن است در افزایش بروز لنفوم نقش داشته باشد. اشکال در روند سیتولیز در سلولهای CD8 و NK cellها با بروز HLH ارتباط دارد. احتمالاً اختلال در نابودی B cellهای مبتلا به EBV در بروز این فنوتیپ نقش داشته باشد (43). نقایص سیتوتوکسیسیتی در سلولهای T و NK با عفونت EBV و بروز HLH همراه بودهاند. پروتئین SLAM نقش مهمی در تکامل T و NK سلها دارد. بیماران XLP1 به خاطر نقص در SLAM استعداد زیادی به عفونت اولیه EBV دارند که این خود منجر به گسترش غیر قابل کنترل پلیکلونال Tو B سلها میشود (44). تنها، بیماران مبتلا به XLP1 مبتلا به لنفوم (30%) میشوند و کولیت هموراژیک مزمن در بیماران XLP2 دیده میشود. محدوده سنی لنفوم در بیماران XLP بین 40-2 سال گزارش شده است. لنفوم ثانویه بندرت در این بیماران گزارش شده و میزان خطر لنفوم در زنان کاریرهنوز مشخص نشده است. اساساً کاریرهای مبتلا به XLP1 بدون علامت هستند ولی در ناقلهای ژن XIAP بیماریهای اریتم ندوزوم، HLH و inflammatory bowel disease (IBD) mild گزارش شده است (45،44). کمبود X- linked inhibitor of (apoptosis (XIAP)که ناشی از موتاسیون در ژن XIAP/BIRC4 میباشد در 2-1 میلیون تولد پسر دیده شده است. مولکول XIAP از طریق عملکرد آنتی آپوپتوتیک خویش در پاسخهای ایمنی اکتسابی نقش ایفا میکند. لنفوسیتهای T بهخصوص invariant natural killer T (iNKT) و mucosal-associated invariant T (MAIT) سلها که سطوح بالای caspas را بیان میکنند، توسط XIAP مهار میشوند. این حالت باعث افزایش حساسیت این سلولها به مرگ سلولی میشود. پروتئین XIAP تنها عضوی از خانواده مهارکنندگان آپوپتوزیس هست که به صورت مستقیم caspasها را مهار میکند (46). لذا تصور میشود که Tسلهای اختصاصی ویروس در بیماران مبتلا به کمبود XIAP عملکرد ساباپتیمالی داشته باشند. دیسرگولیشن ایمنی در بیماران مبتلا به XIAP deficiency تظاهرات بالینی متنوعی را بهوجود میآورد که شامل: HLH مکررکه اغلب توسط EBV ایجاد میشود، IBD، اسپلنومگالی، هیپوگاماگلوبولینمی، سیتوپنی و پدیدههای اتوایمیونیتی میباشد (47). مهمترین فنوتیپ بالینی بیماران مبتلا بهXIAP deficiency استعداد به HLH در زمینه عفونت EBVاست. بیماری HLH نسبت به بیماران XLP1 شدت کمتری دارد. بیش از نیمی از بیماران با XIAP deficiency هرگز عفونت EBVرا تجربه نمیکنند. همچنین این افراد در مقایسه با XLP2 مبتلا به لنفوم نمیشوند. احتمالاً به خاطر خاصیت آنتیآپوپتوتیک XIAP که بیماران را از بروز کانسر محفوظ نگه میدارد. اخیرا به همین جهت XIAP به عنوان یک هدف امیدوار کننده در در مان سرطان مورد توجه قرار گرفته است (48).
IL-2–inducible T-cell kinase deficiency (ITK): بیماری ITK deficiency در واقع یک نقص ایمنی ترکیبی شدید است با توارث اتوزومال مغلوب که فرد را مستعد به HLH و لنفوم هوچکین میکند (49). مولکول ITK از دسته پروتئینکینازهای غیر رسپتوری خانواده TEC هست که روی T cell، NK cell، iNK cell و ماست سلها قرار دارد. بیمارانی که تا کنون گزارش شدهاند با عوارض مرتبط با عفونت EBV مانند بیماریهای لنفوپرولیفراتیو و بدخیمیهای مرتبط با این ویروس بودهاند (50). خصوصیات ایمونولوژیک بیماران با ITK deficiency شامل هیپوگاماگلوبولینمیای پیشرونده در کنار لنفوپنی و کاهش پیشرونده CD4+ Tcellها میباشد. همانند سایر افراد مبتلا به بیماری لنفوپرولیفراتیو وابسته به EBV، بیماران ITK deficient دارای سطوح شدیداً کاهش یافته NKT cell ها در خون محیطی هستند که نقش کلیدی در دفاع علیه ویروس EBV به عهده دارند (51). بر خلاف بیماران مبتلا به XLP1 که مستعد لنفوم بورکیت هستند این بیماران بیشتر با هوچکین مراجعه میکنند.
Autoimmune Lymphoprolifrative Syndrome (ALPS): سندرم لنفوپرولیفراتیو اتوایمیون یک اختلال ژنتیکی نادریست با زمینه ژنتیکی پیچیده که علاوه بر مشخصههای ژنتیکی مختلف الگوی توارثی آن نیز متفاوت میباشد. الگوهای توارثی اتوزومال غالب، اتوزومال مغلوب ومواردی موتاسیونهای سوماتیک نیز در این بیماران گزارش شده است. تظاهرات بالینی آن شامل: لنفادنوپاتی، ارگانومگالی، رویدادهای اتوایمیونیتی بهخصوص اتوایمیون سایتوپنی و لنفوم میباشد. هال مارک بیماری فنوتیپ ایمونولوژیک بهخصوصی است به صورت افزایشTCR α/β CD4-CD8- “double negative” T cellها. سایر بیومارکرهای مشاهده شده در ALPS شامل افزایش سطوح ویتامین B12، IL-10 ، soluble FAS ligand میباشد. در این بیماران اختلال در آپوپتوزیس وجود دارد که با واسطه FAS میباشد (53،52). علائم بالینی بیماران طیف وسیعی را شامل میشود که نتیجه هیپرپلازی غیر بدخیم لنفوییدی و تجمع پیشرونده سلولهای Auto reactive لنفوسیتهای T و B میباشد. میزان نفوذ علائم بالینی در بیماران بهطور کامل مشخص نشده است و بستگی به محل و نوع موتاسیون ژنتیکی دارد و حتی ممکن است در افرادی که موتاسیون مشابهی دارند علائم متفاوت باشد. اتوایمیون سایتوپنی شایعترین علامتی است که تمام ردههای خونی را میتواند درگیر کند. درگیری ارگانهای توپر شایع نیست و مواردیکه گزارش شده شامل گلومرولونفریت، هپاتیت، بافت همبند (لوپوس)، یووئیت، تیروئیدیت و گیلنباره بوده است. در مطالعهای که توسط Azizi و همکارانش روی 780 بیمار ALPS و ALPS-like syndrome صورت گرفت شایعترین تظاهر بالینی اسپلنومگالی (92/5%) و لنفادنوپاتی (86/8%) بود. اما در گروه ALPS-like بیماریهای هماتولوژیک (87/8%) و اتوایمیون (86/8%) شایعتر بود. در این بیماران بدخیمی به فرمهای متفاوتی وجود داشت. لنفوم هوچکین در 25 بیمار و لنفوم نانهوچکین در 26 بیمار گزارش شد. میزان بدخیمی بین دو گروه تفاوت معنیداری نداشت (54). بیماران باgerm line mutation در قسمت داخل سلولی پروتئین FAS ریسک بالاتری برای بروز لنفوم هوچکین و نان هوچکین دارند. در مطالعهای خطر نسبی لنفوم هوچکین و لنفوم نان هوچکین به ترتیب 51 و 14 بود. بیماران مبتلا به ALPS لنفادنوپاتی مزمن نوساندار دارند. بنابراین باید این افراد بهصورت مرتب جهت تشخیص زودهنگام لنفوم، توسط CT scan سریال و scan PET تحت مانیتورینگ قرار بگیرند (55).
نقایص سیستم ایمنی ذاتی
WHIM syndrome: سندرم WHIM یک نقص ایمنی نادری است که با حروف اختصاری کلمات Wart-hypogammaglobulinemia-Infection-myelokathexis تعریف میشود. میلوکاتکسی یک فرم غیر معمول از نوتروپنی مادرزادی غیرسیکلیک است که به علت تجمع نوتروفیلهای بالغ و دژنره در مغز استخوان ایجاد میشود. معمولاً مونوسیتوپنی و لنفوپنی بهخصوص B cell lymphopenia هم در این افراد دیده میشود. این بیماری توارث اتوزومال غالب دارد که ناشی از موتاسیون gain of function در ژن کدکننده G protein couple recetor(CXCR4) میباشد (56). اکثریت بیماران پان لکوسیتوپنی، نوتروپنی، لنفوپنی همراه با هیپوگاماگلوبولینمیا دارند. مطالعات، تنوع وسیعی را در فنوتیپ بالینی بیماران نشان داده است. انسیدانس متغیر در عفونتهای مکرر (عفونت تنفسی-عفونت پوستی - بیماری پریدونتال- استئومیلیت و مننژیت)، استعداد به عفونتهای HPV و EBV، نقایص کونوترانکال قلب و افزایش استعداد به بدخیمیها از جمله این علائم هستند. در کل تشخیص این بیماری بهصورت کلینیکوپاتولوژیکال است. اطلاعات کافی در مورد سیر طبیعی بیماری در بیماران وجود ندارد اما بعضی عوارض بیماری مثل برونشکتازی، کری و بدخیمیها را ممکن است با تشخیص زودرس بیماری پیشگیری کرد (57). این بیماران مستعد عفونت HPV هستند که باعث ایجاد زگیل در مخاط دهان-ناحیه تناسلی و پوست میشود و این حالت خود باعث افزایش ریسک اسکواموس کارسینوما میگردد. در یک مطالعه آنالیزی روی 18 بیمار مبتلا به سندرم WHIM صورت گرفت. انسیدانس زگیل در 61% بیماران و میزان بروز بدخیمیهای مرتبط با HPV 16 % گزارش شد (58). نوتروپنی مادرزادی (congenital neutropenia): نوتروپنی مادرزادی در برگیرنده گروهی از اختلالات است که با نوتروپنی پایدار یا متناوب شدید یا خفیف مشخص میشود. فیزیولوژیوپاتولوژی نوتروپنی در این بیماران هنوز بهطور دقیق مشخص نیست. ولی توقف تولید نوتروفیلها در مغز استخوان اثبات شده است. یک زنجیرهای بین نوتروپنی پایدار در مقابل نوتروپنی گذرا و موتاسیونها مختلف در لوکوس 3. 13 p 19 ژن کدکننده آنزیم نوتروفیل الاستاز (ELANE) وجود دارد. مکانیسمهای مولکولی که بهوسیله آن موتاسیونهای ELANE باعث اختلال در میلوپوئزیس (Myelopoiesis) میشود ناشناخته است. نوتروپنی مادرزادی شدید یک فرم نارسایی مغز استخوان است که بیمار از بدو تولد دارای نوتروپنی شدید همراه با استعداد به عفونتهای مختلف میباشد. نوتروپنی مرتبط با ELANE، سیکلیک نوتروپنی و نوتروپنی مادرزادی شدید (سندرم کاستمن) است که تقریباً فنوتیپ بالینی مشابهی دارند. موتاسیونهای EALNE در 100%-80% بیماران سیکلیک نوتروپنی و در 63%-35% موارد نوتروپنی مادرزادی شدید یافت میشود (59). بیماران مبتلا به severe congenital neutronpenia(SCN) دارای نوتروفیلهای خون محیطی کمتر از µ/200 هستند و اغلب همراه با سطوح افزایش یافته مونوسیتها میباشند. بیشترین موتاسیونی که در بیماران با ازدواجهای غیر فامیلی دیده میشود در ژن ELANE است در حالیکه میزان موتاسیون در ژن HAX1 در جمعیتهای با ازدواجهای فامیلی بیشتر دیده میشود. سابتایپهای SCN براسای الگوی توارثی شان و موتاسیون در ژنهای مختلف مشخص میشوند مثل موتاسیون در ژنهایSCF3R- JAGN1- VPS45- G6PC3- HAX1 که الگوی توارثی اتوزومال مغلوب دارند. ژنهای GFI1- ELANE- SRP54 توارث اتوزومال غالب و ژن WAS وابسته به X دارند (60). موتاسیونهای ELAN شایعترین علت SCN هستند که تقریباً 50% موارد را به خود اختصاص میدهند. در 30% موارد، علت ژنتیکی SCN ناشناخته است. علت مهم مورتالیتی در بیماران با SCN از زمان مصرف گسترده G-CSF، میلودیسپلازی و لوکمی میلوسیتیک حاد (AML/MDS) میباشد. رجیستری بیماران نوتروپنی در فرانسه، میزان بروز تجمعی AML یا MDS را 8/10% طی 20 سال در بیماران SCN گزارش کرده است. در مطالعه آیندهنگر دیگری روی 374 فرد مبتلا به SCN که به مدت طولانی تحت درمان G-CSF بودند بروز تجمعی AML/MDS بعد از 15 سال 22% اعلام شد. بیمارانی که به دوزهای بالاتر G-CSF نیاز داشتند و پاسخ ضعیفتری نشان میدادند در معرض ریسک بیشتری برای لوکمی بودند. لازم به ذکر است که AML وMDS در موتاسیونهایELAN- HAX1- G6PC3-WAS هم گزارش شده است (61).
نتیجهگیری
بیماران مبتلا به نقص سیستم ایمنی از آنجاییکه مستعد عفونتهای متعدد میباشند این قابلیت را دارند که بهدنبال برخی ویروسها و عوامل کارسینوژن دچار بدخیمیهای گوناگون بشوند (جدول 1). این افراد همچنین در بعضی موارد دارای اتوایمیونیتی و دیسرگولیشن ایمنی هستند که این خطرات را افزایش میدهد. از طرف دیگر وجود و تکرار بعضی بدخیمیها در خانوادهها به خصوص بدخیمیهای خونی این هشدار را به ما میدهد که ممکن است در پس این بدخیمیها، اختلالات سیستم ایمنی از جمله نقایص ایمنی اولیه نهفته باشد و لزوم ارزیابی سیستم ایمنی را در گروهی از این بیماران ایجاب میکند.
حامی مالی: ندارد
تعارض در منافع: وجود ندارد.
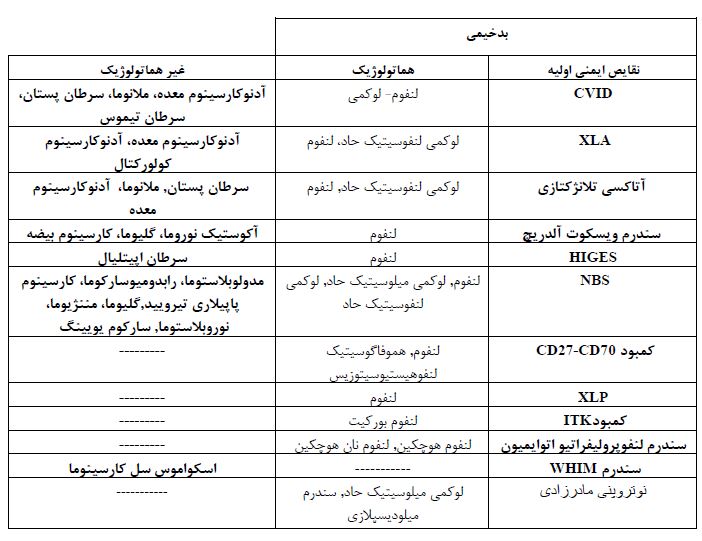
جدول 1 : بدخیمیهای هماتولوژیک و غیر هماتولوژیک در بیماران مبتلا به نقص ایمنی اولیه
نقایص ایمنی اولیه اختلالات نسبتاً نادر و هتروژنی هستند که یک یا چندین عضو از سیستم ایمنی دچار اختلال عملکرد میشود. تظاهرات بالینی افراد مبتلا به این بیماریها بسیار متغیر و متنوع هستند. اکثریت این افراد با توجه به نوع اختلالی که دارند دچار عفونتهای مکرر با جرمهای مختلف میشوند. این افراد به دلایل مختلفی ممکن است مستعد بیماریهای بدخیم باشند (1). نوع بدخیمی بستگی دارد به نوع نقص ایمنی اولیه، سن بیمار و نوع ویروسی که بیمار به آن مبتلا میشود. لذا مکانیسمهای پاتوژنیک مختلفی در بروز این بدخیمیها نقش دارند. تصور میشود پاسخ ایمونولوژیک میزبان نسبت به تحریک مزمن آنتیژنیک در بروز کانسر مهمترین نقش را داشته باشد (2). پیشرفتهای بسیار مهمی در خصوص درک مکانیسمهای مولکولی نقایص سیستم ایمنی سلولار و هومورال صورت گرفته است که دانستههای ما را درباره بروز انواع عفونتها و تومورها درهریک از این اختلالات افزایش داده و در تشخیص زودهنگام این عوارض به ما کمک میکند (3). شواهد متعددی وجود دارد که نشان میدهد سیستم ایمنی در بروز بسیاری از بدخیمیها نقش مهمی دارد. از اینرو نقایص ایمنی از دسته اختلالاتی هستند که ممکن است فرد را به کانسر مبتلا کنند. مکانیسمهای متعددی در بروز بدخیمی در بیماران مبتلا به نقص ایمنی اولیه دخالت دارند: ازجمله نقصهایی در آپوپتوزیس و تمایز سلولی مانند اختلالات سیتواسکلتون، متابولیسم سلولی، سیتوتوکسیسیتی. همچنین ناهنجاریهایی در ماده ژنتیکی سلول مثل ناپایداری کروموزومی، و اشکال در ترمیم DNA نیز در ایجاد کانسر دخالت دارند. از سوی دیگر عوامل خارجی مثل عفونت با ویروسهای انکوژن به عنوان مثال (Epstein-Barr virus) EBV و (Human papilloma virus)HPV و همچنین وجود التهاب بافتی مزمن، فاکتورهایی هستند که فرد مبتلا به نقص ایمنی را مستعد بدخیمی خواهند کرد (4). تقریباً در10% سرطانها استعداد خانوادگی مشاهده میشود. این در حالی است که در 80%-60% سرطانهای خانوادگی بررسی ژنتیکی صورت نمیگیرد. در مطالعهای که روی 3658 بیمار مبتلا به نقص ایمنی اولیه بین سالهای 2015-2003 صورت گرفته بود میزان خطر نسبی سرطان در مقایسه با جمعیت عمومی 1/24 گزارش شد. در جنس مذکر میزان این خطر 1/19بود ولی در جنس مونث میزان خطر نسبی در گروه نقایص ایمنی اولیه و جمعیت عمومی یکسان بود (5). در برخی خانوادهها اتوایمیونیتی و استعداد به عفونت دیده میشود. در سالهای اخیر رابطه اثبات شدهای بین دیس رگولیشن سیستم ایمنی و کانسر گزارش شده است. بنابراین جای تعجبی ندارد که بیماران دارای اختلال سیستم ایمنی مستعد به کانسر باشند (6). ما در این مطالعه قصد داریم مروری کوتاه بر جنبههای اپیدمیولوژیک، پاتوفیزیولوژیک و بالینی نقایص سیستم ایمنی اولیه داشته باشیم که انواع مختلف بدخیمی در آنها بوجود میآیند.
روشبررسی
جستجوی مقالات در پایگاه های اطلاعاتی google scholar،pubmed، web of science ،scopus صورت گرفت. در این بررسی سعی شده است از جدیدترین و معتبرترین مقالات مرتبط با موضوع که به زبان انگلیسی به چاپ رسیده است استفاده شود. برای انسجام و نظم بیشتر مطالب سعی شده عناوین مورد بحث براساس تقسیم بندی IUIS 2023 مطرح شود.
Humoral immunedeficiency
(common variable immunodeficiency)(CVID): شایعترین نقص ایمنی هومورال اولیه علامتدار بیماری (CVID) است. این بیماری گروهی از اختلالات هتروژن را در برمیگیرد که در تولید آنتی بادی نقص دارند. شایعترین تظاهر بالینی بیماران مبتلا به CVID عفونتهای سینوپولمونری و گوارشی میباشد. باکتریهایی مثل گونههای استرپتوکک، هموفیلوس آنفولانزا، موراکسلا کاتارالیس، نایسریا و گونههای استافیلوکک میگروارگانیسمهای شایع در این افراد هستند. همچنین پاتوژنهایی مثل مایکوپلاسما، رینوویروسها، سیتومگالوویروس و پنوموسیستیس ژیروویسی اگر چه در این افراد شایع نیست ولی گهگاهی دیده میشوند (7). این بیماران در کنار استعداد به عفونتهای مختلف مستعد یکسری بیماریهای غیر عفونی نیز میباشند. مثل: بیماریهای خود ایمنی، اختلالات اتواینفلیمیتوری و اختلالات لنفوپرولیفراتیو پلی کلونال. میزان پریواالانس بدخیمی در این نوع اختلال حدود 8/6% تخمین زده میشود (8). این بیماران 47- 7 برابر شانس بروز کانسر نسبت به جمعیت عمومی دارند. لنفوم (40/5%) و آدنوکارسینوم معده (7/5%) شایعترین بدخیمیهای گزارش شده در بیماران مبتلا به CVID هستند. سایر بدخیمیها مثل کانسر پستان، پوست، تیموس، لوکمی، و ملانوم در ردههای بعدی قرار دارند. کانسر معده اولین علت مرگ مرتبط با بدخیمی در بین بیماران CVID میباشد. این در حالیست که در جمعیت عمومی پنجمین علت مرگ به حساب میآید (13-9). میزان بروز سرطان در بیماران مبتلا، در دهههای چهارم تا ششم بیشترین مقدار است. مکانیسمهای پاتوفیزیولوژیک ایجاد بدخیمی در بیماران مبتلا به CVID به طور کامل مشخص نشده است. اما تصور میشود مکانیسمهایی چون ناپایداری و استعداد ژنتیکی، فعالیت و تکثیر مداوم و افزایش یافته سلولهای لنفوییدی طی دورههای عفونت و اختلال در کلیرانس ویروسهای اونکوژنیک و عفونتهای باکتریال در ایجاد بدخیمی نقش داشته باشند. سلولهای کلیدی در کنترل سلولهای تومورال، NK cell ها و لنفوسیتهای CD8+ سیتوتوکسیک هستند. همچنین سلولهای CD4+ هم بهخصوص TH1 cellها با کمک به فعالشدن CD8+سلها در کنترل سلولهای بدخیم میتوانند نقش ایفا کنند. اختلال درپاسخهای ایمونولوژیک سلولهای CD4+ وCD8+ و همچنین کاهش سلولهای (Natural killer cell) NK cell در بیماران(Common variable immunodeficiency) CVID میتوانند توجیهکننده بروز بیماریهای بدخیم باشند (14).
(X-linked agammaglobulinemia)(XLA): آگاماگلوبولینمیای وابسته به X برای اولین بار در سال 1952 توسط شخصی بنام بروتون تشخیص داده شد. دراین افراد بهدلیل اختلال در تمایز پیشسازهای لنفوسیت B در مغز استخوان، لنفوسیتهای B خون محیطی و پلاسماسلها به شدت کاهش یافته هستند و به تبع آن تمام ردههای ایمونوگلوبولینها افت شدید دارند. فرمهای اوتوزومال مغلوب بیماری نیز گزارش شده است که حاصل موتاسیون در ژنهای µ heavy chain، Igα، Igβ،λ5 ، BLNK، PIK3R1 میباشند (16،15،7). در مطالعهای که توسط آقامحمدی و همکاران صورت گرفته بود تظاهرات بالینی، یافتههای ایمونولوژیک و عوارض در بیماریXLA و CVID مورد مقایسه قرار گرفتند و مشخص شد همه موارد بدخیمی در XLA نسبت به CVID کمتر میباشد (17). علی رغم اینکه بدخیمیها در بیماران XLA نادر است اما کیس ریپورتهایی در مورد وجود آدنوکارسینوم معده که احتمالاً به علت گاستریت مزمن ناشی از هلیکوباکتر پیلوری میباشد گزارش شده است. سایر بدخیمیهایی که در این افراد مشاهده شده شامل(Acute lymphocytic leukemia ) ALL – لنفوما – آدنوکارسینوم کولورکتال میباشد ( 20-18).
(cellular and combined immunodeficiency)
Ataxia telangiectasia: اتاکسی تلانژکتازی یک بیماری نادر و مولتیسیستمی است با توارث اتوزومال مغلوب که با موتاسیون در ژنataxia telangiectasia mutated gene(ATM) مشخص میشود. محصول این ژن پروتئینکینازی است که در کنترل سیکل سلولی و ترمیم مولکول DNA دخالت دارد. فرم کلاسیک بیماری بهصورت شروع زودرس آتاکسی مخچهای، تلانژکتازی، عفونت مکرر ناشی از نقص ایمنی، رادیوسنسیتیویتی، استعداد به بدخیمی و افزایش آلفافیتوپروتئین تظاهر مییابد. اکثریت بیماران در دهه اول بیماری وابسته به ویلچر میشوند و در دهه دوم زندگی به دلیل نارسایی تنفسی یا بدخیمی فوت خواهند کرد (22،21). در این بیماران ناپایداری کروموزومی ناشی از اختلال در ژن ATM وجود دارد که فرد را مستعد رادیوسنسیتیویتی و بدخیمی میکند. حتی کاریرهای هتروزیگوس ژن ATM نیز به خاطر استعداد به بدخیمی کاهش امید به زندگی دارند. آتاکسی تلانژکتازی به دو فرم تظاهر مییابد. یکی فرم کلاسیک و شدید که تظاهرات بالینی زودرس در دوران کودکی دارند. وارینت دیگری هم وجود دارد که خفیفتر است و تظاهرات بیماری در سنین نوجوانی و بزرگسالی بروز پیدا میکند. در مطالعهای که توسط Emily Petley و همکارانش صورت گرفت 1826 بدخیمی را در 1643 بیمار مبتلا به اتاکسی تلانژکتازی مورد بررسی قرار دادند. شایعترین بدخیمی در گروه کلاسیک، نانهوچکین لنفوما با سن متوسط 9 سال و 8 ماه بود. دومین بدخیمی لوکمی لنفوسیتیک حاد با سن متوسط 11 سال بود و در رده سوم لنفوم هوچکین قرار داشت. در گروه وارینت شایعترین کانسر سرطان پستان و بهدنبال آن لوکمی گزارش شد (23). بر اساس مطالعات مختلف شایعترین بدخیمیها در این افراد بدخیمیهای هماتولوژیک به خصوص فرمهای لنفوییدی است. اما برخی solid tumorها مانند سرطان معده، پستان وپوست (ملانوما) هم در این بیماران گزارش شده است (24).
(Wiscott – Aldrich syndrome)(WSA): سندرم ویسکوت آلدریچ یک بیماری نادر وابسته به x است که در سال 1937 میلادی گزارش شد. بیماران با تریاد میکروترومبوسیتوپنی، عفونت مکرر و اگزما تظاهر پیدا میکنند. این افراد همچنین مستعد خودایمنی و بدخیمیهای لنفوییدی نیز هستند. ژن WAS روی بازوی کوتاه کروموزوم xقرار گرفته و پروتئینی را به نام WASP در سلولهای بنیادین مغز استخوان، رده لنفوییدی و میلوییدی کد میکند. نقش این پروتئین در پلیمریزاسیون اکتین و بازسازی اسکلت سلولی است. همچنین تنظیمکننده سیناپس ایمونولوژیک سلولهای B وT نیز میباشد. از آنجاییکه WASP انحصارا روی سلولهای هماتوپوئتیک اکسپرس میشود، لذا بیماران مبتلا به ویسکوتآلدریچ همراه با اختلالاتی در سیستم ایمنی و هماتوپوئتیک هستند. پروتئین WASP توسطWIP (WASP-interacting-protein) تنظیم میشود. مطالعات نشان داده است لنفوسیتهای T در بیماران مبتلا به ویسکوت آلدریچ و موشهایی که کمبود WASP و WIP دارند درT cell receptor signaling اشکال دارند. شواهد نشان میدهد که این اختلال در پاتوژنز لنفوم T cell نقش مهمی دارد. این بیماران مستعد عفونتهای سینوپولمونری، ویروسی (هرپس و واریسلا)، عفونتهای کاندیدایی و عفونتهای فرصتطلب مثل پنوموسیستیس ژیروویسی هستند. شایعترین بدخیمی در این افراد سرطانهای هماتولنفوییدی است. این بدخیمیها در سنین نوجوانی و بزرگسالی شیوع بیشتری دارند تا در کودکان. بدخیمیهای ناشایعترکه در این افراد گزارش شده است شامل گلیوما، آکوستیک نوروما و کارسینوم بیضه بوده است (27-25). در مطالعهای perry و همکارانش خطر نسبی (Relative risk) کانسر را در بیماران مبتلا 100 برابر جمعیت عمومی گزارش کردند که با افزایش سن، میزان خطر افزایش می یافت (28). در مطالعه دیگری که Sukru Cekic و همکارانش روی 6392 بیمار مبتلا به نقص ایمنی انجام دادند 59 بیمار همراه با بدخیمی را مورد ارزیابی قرار دادند. فرکانس بدخیمی در ویسکوتالدریچ 9/1% گزارش شد (29).
Hyper IgE syndrome: سندرم hyper IgE جزء گروهی از خطاهای ارثی ایمنی است که با استعداد به عفونتهای ریوی و پوستی، اگزما و افزایش شدید سطح سرمی IgE مشخص میشوند. فرم کلاسیک آن ناشی از موتاسیون در ژن کدکننده STAT3 (signal transduction and activator of transcription 3) میباشد. این بیماران مستعد عفونتهای استافیلوککی پوستی و ریوی هستند. عفونتهای کاندیدایی هم در این بیماران به دلیل اختلال در محورIL17-TH17 مشاهده میشود. تظاهرات غیر عفونی این بیماران شامل آلرژی، اختلالات بافت همبند (صورت خشن، پل بینی پهن، تاخیر در افتادن دندانهای شیری، شکنندگی استخوانها، هیپراکستنسی بیلیتی (Hyper extensibility) مفاصل و...)، اختلالات عروقی، خودایمنی و بالاخره استعداد به بدخیمی میباشد. 7% بیماران مبتلا به موتاسیون ژن STAT3 دچار بدخیمی میشوند که شایعترین فرم آن بیماری لنفوم میباشد (30). فرم اتوزومال مغلوب یماری hyper IgE بیماری DOCK8 deficiency است. آتوپی (آلرژی غذایی- درماتیت و آسم)، عفونتهای سینوپولموناری و عفونتهای ویروسی در پوست، بدخیمیهای زودرس، افزایش IgE و ائوزینوفیلی خون محیطی از جمله علائم شایع در این افراد است (31). مطالعهای توسط Susanne E روی 136 بیمار مبتلا به DOCK8 صورت گرفت. میزان بدخیمی در این بیماران 17% گزارش شد. شایعترین بدخیمی انواع هماتولوژیک بود (48%)، کانسرهای اپیتلیال (39%) و سایر بدخیمیها (22%) در ردههای بعدی قرار داشتند (32).
Nijmegen breakage syndrome (NBS): سندرم NBS یک بیماری نادر اتوزومال مغلوب است. ناپایداری کروموزومی مشخصه بارز بیماری به حساب میآید. شیوع بیماری در نواحی اروپای مرکزی و اروپای شرقی بیشتراست (لهستان، جنوب آلمان، جمهوری چک، اوکراین). موارد اسپورادیک هم در خاور میانه گزارش شده است (33). این افراد میکروسفالی بدو تولد همراه با اختلال ساختاری صورت به شکل bird-like و کاهش ضریب هوشی بدون سایر تظاهرات نورولوژیک دارند. سایر علائم آنها شامل استعداد به عفونتهای مکرر، اختلال رشد خفیف، نارسایی زودرس تخمدانها و تمایل شدید به بیماریهای بدخیمی بهخصوص بدخیمیهای خونی میباشد. رادیوسنسیتیویتی و ناپایداری کروموزومی از خصوصیات بارز این بیماران است. وجود موتاسیون در هر دو آلل ژن nibrin (NBN) تشخیص NBS را کامل میکند (34). نقص ایمنی در این بیماران به شکل کاهش سلولهای Tو B(80%)، هیپوگاماگلوبولینمیای شدید (20%) و IgA deficiency (50%) میباشد. بدخیمی علت مهم مرگ و میر در این افراد است. بیش از 40% بیماران تا سن 20 سالگی مبتلا به کانسر میشوند که اغلب منشا لنفوییدی دارد (diffuse large B cell lymphoma،T cell lymphoblastic lymphoma). سایر بدخیمیهای هماتولوژیک در این افراد شامل هوچکین،ALL T & B cell و AML میباشد. تومورهای solid درNBS نیز بهصورت نادر دیده میشوند که عبارتند از مدولوبلاستوما، رابدومیوسارکوما، کارسینوم پاپیلاری تیروئید، گلیوما، مننژیوما، نوروبلاستوما و یوئینگسارکوما (35). تحقیقی توسط Svetlana. O روی 136 بیمار مبتلا به NBS انجام شد. بدخیمی در 45/6% افراد مورد بررسی گزارش شد. نسبت مرد به زن 1/5 به 1 بود. نئوپلاسمهای لنفوییدی اکثریت بدخیمیها را به خود اختصاص داد (9/91%). لنفوم نان هوچکین شایعترین بدخیمی بود که 2/3 موارد را شامل میشد. در حالیکه لنفوم هوچکین با فرکانس 3/3% فرم نادرتری در NBS بود. تومورهای solid فقط 5/6% کل نئوپلاسمها را در بر میگرفت (33).
Diseases of immune dysregulation
CD27-CD70 deficiency: مولکولهای CD70 و CD27 متعلق به خانواده بزرگ رسپتورهای TNFمیباشند و نقش مهمی در ایجاد و حفظ ایمنی سلولی دارند. این مسیر ایمونولوژیک در ایمنی ضد ویروسی نقش مهمی دارد. اخیراً مشخص شده که اختلال در این مولکولها فرد را مستعد عوارض لنفوپرولیفراتیو ویروس EBV میکند. بیماران مبتلا به کمبود CD27 استعداد بسیار زیادی به اختلالات لنفوپرولیفراتیو ناشی از عفونت EBV دارند (37،36). مولکول CD70 یک لیگاند کمک تحریکی (co-stimulatory) است که روی تعدادی از سلولهای ایمونولوژیک مانند T cellها وB cellها و سلولهای دندریتیک بیان میشود. اتصال این مولکول با رسپتورش، CD27، باعث فعال شدن سلولهای ایمونولوژیک CD27+ شده میزان بقا و تکثیر آنها را افزایش میدهد. تعامل بین CD27/CD70 برای تکامل B cellها الزامی نیست اما به نظر میرسد که در فعال شدن B cellها، تولید آنتیبادی و تشکیل ژرمینال سنتر نقش مهمی داشته باشد. یافتههای بالینی و ایمونولوژیک بیمارانی که در محور CD27-CD70 اختلال دارند شامل وجود نقص ایمنی اولیه با دیسرگولیشن سیستم ایمنی، اشکال در تکامل B cellهای ترمینال، اتوایمیونیتی، عفونتهای ویروسی شدید و لنفوما میباشد (39،38،4). شایعترین تظاهرات بالینی بیماران با اختلال محور CD27-CD70 وابسته به عفونت EBV است. در مطالعهای که توسط Sujal Ghosh روی 49 بیمار صورت گرفت 37% بیماران اختلال لنفوپرولیفراتیو، 43% لنفوما و 18% HLH داشتند. تست مثبت EBV در بدو تشخیص، در 31 فرد از 33 بیمار مبتلا به CD27 deficiency و 15 نفر از 16 بیمار مبتلا به CD70 deficency گزارش شد (40). یافتههای بالینی هیپوگاماگلوبولینمیا در بیماران با کمبود CD27 و CD70 ناشی از دیسرگولیشن سیستم ایمنی با واسطه ویروس EBV میباشد. این بیماران میتوانند با مونوکلئوزیس شدید – هموفاگوسیتیک لنفوهیستیوسیتوزیس (HLH) – اختلالات لنفوپرولیفراتیو و لنفوم تظاهر یابند. در مطالعهای 11 بیمار از 21 بیمار مبتلا به CD70- deficiency و 36% (12/33) از بیماران CD27-deficient دچار لنفوم شده بودند و شایعترین فرم آن لنفوم هوچکین بود. مکانیسمهای دقیق این بینظمیهای ایمونولوژیک بهطور کامل مشخص نیست اما به نظر میرسد که CD27 و CD70 در کنترل ایمونولوژیک بدخیمیها جدای از وجود عفونت EBV نقش مهمی دارند (41).
(سندرم دونکان)x-linked lymphoproliferative syndrome(XLP): بیماری XLP یک نقص ایمنی اولیه نادری است که با استعداد زیاد به عفونت EBV – دیس گاماگلوبولینمی – اختلالات لنفوپرولیفراتیو و هموفاگوسیتیک لنفوهیستیوسیتوزیس تظاهر پیدا میکند. این بیماری برای اولین بار در سال 1970 گزارش شد. در حالیکه اولین ژن مسبب بیماری کهSH2D1A میباشد و کد کنندهSLAM- associated protein ((SAP است، در سال 1998 کشف شد. دومین ژن بیماری XIAP در سال 2006 در بیمارانی پیدا شد که علائم شبیه XLPداشتند ولی موتاسیون درژن SH2D1A وجود نداشت. بنابراین XLP به دو دسته تقسیم شد: 1- SAP- deficiency(XLP1) که موتاسیون در ژن SH2D1A وجود دارد. 2-XIAP- deficiency(XLP2) (42). مولکول SAP تنظیمکننده فعالیت B cellها و همچنین تنظیمکننده فعالیت لیتیک T cell ها میباشد. فعالیت پروآپوپتوتیک SAP باعث جلوگیری از تکثیر سلولهای آسیب دیده میشود که این خود در پیشگیری از ایجاد کانسر نقش کلیدی دارد. از طرفی اشکال در بقاء سلولهای خونساز ناشی از اختلال در پاسخهای سیتوتوکسیک T و NKسلها با واسطه رسپتورSLAM، ممکن است منجر به بروز بدخیمی گردد. نقص SAP در سلولهای CD8 منجر به اختلال در چسبندگی و نابودی B cell های فعال شده بهویژه B cellهای تغییر ماهیت یافته توسط EBV میشود که سطوح بالای SLAM را بیان میکنند. اختلال در بقا ایمونولوژیک B cell ممکن است در افزایش بروز لنفوم نقش داشته باشد. اشکال در روند سیتولیز در سلولهای CD8 و NK cellها با بروز HLH ارتباط دارد. احتمالاً اختلال در نابودی B cellهای مبتلا به EBV در بروز این فنوتیپ نقش داشته باشد (43). نقایص سیتوتوکسیسیتی در سلولهای T و NK با عفونت EBV و بروز HLH همراه بودهاند. پروتئین SLAM نقش مهمی در تکامل T و NK سلها دارد. بیماران XLP1 به خاطر نقص در SLAM استعداد زیادی به عفونت اولیه EBV دارند که این خود منجر به گسترش غیر قابل کنترل پلیکلونال Tو B سلها میشود (44). تنها، بیماران مبتلا به XLP1 مبتلا به لنفوم (30%) میشوند و کولیت هموراژیک مزمن در بیماران XLP2 دیده میشود. محدوده سنی لنفوم در بیماران XLP بین 40-2 سال گزارش شده است. لنفوم ثانویه بندرت در این بیماران گزارش شده و میزان خطر لنفوم در زنان کاریرهنوز مشخص نشده است. اساساً کاریرهای مبتلا به XLP1 بدون علامت هستند ولی در ناقلهای ژن XIAP بیماریهای اریتم ندوزوم، HLH و inflammatory bowel disease (IBD) mild گزارش شده است (45،44). کمبود X- linked inhibitor of (apoptosis (XIAP)که ناشی از موتاسیون در ژن XIAP/BIRC4 میباشد در 2-1 میلیون تولد پسر دیده شده است. مولکول XIAP از طریق عملکرد آنتی آپوپتوتیک خویش در پاسخهای ایمنی اکتسابی نقش ایفا میکند. لنفوسیتهای T بهخصوص invariant natural killer T (iNKT) و mucosal-associated invariant T (MAIT) سلها که سطوح بالای caspas را بیان میکنند، توسط XIAP مهار میشوند. این حالت باعث افزایش حساسیت این سلولها به مرگ سلولی میشود. پروتئین XIAP تنها عضوی از خانواده مهارکنندگان آپوپتوزیس هست که به صورت مستقیم caspasها را مهار میکند (46). لذا تصور میشود که Tسلهای اختصاصی ویروس در بیماران مبتلا به کمبود XIAP عملکرد ساباپتیمالی داشته باشند. دیسرگولیشن ایمنی در بیماران مبتلا به XIAP deficiency تظاهرات بالینی متنوعی را بهوجود میآورد که شامل: HLH مکررکه اغلب توسط EBV ایجاد میشود، IBD، اسپلنومگالی، هیپوگاماگلوبولینمی، سیتوپنی و پدیدههای اتوایمیونیتی میباشد (47). مهمترین فنوتیپ بالینی بیماران مبتلا بهXIAP deficiency استعداد به HLH در زمینه عفونت EBVاست. بیماری HLH نسبت به بیماران XLP1 شدت کمتری دارد. بیش از نیمی از بیماران با XIAP deficiency هرگز عفونت EBVرا تجربه نمیکنند. همچنین این افراد در مقایسه با XLP2 مبتلا به لنفوم نمیشوند. احتمالاً به خاطر خاصیت آنتیآپوپتوتیک XIAP که بیماران را از بروز کانسر محفوظ نگه میدارد. اخیرا به همین جهت XIAP به عنوان یک هدف امیدوار کننده در در مان سرطان مورد توجه قرار گرفته است (48).
IL-2–inducible T-cell kinase deficiency (ITK): بیماری ITK deficiency در واقع یک نقص ایمنی ترکیبی شدید است با توارث اتوزومال مغلوب که فرد را مستعد به HLH و لنفوم هوچکین میکند (49). مولکول ITK از دسته پروتئینکینازهای غیر رسپتوری خانواده TEC هست که روی T cell، NK cell، iNK cell و ماست سلها قرار دارد. بیمارانی که تا کنون گزارش شدهاند با عوارض مرتبط با عفونت EBV مانند بیماریهای لنفوپرولیفراتیو و بدخیمیهای مرتبط با این ویروس بودهاند (50). خصوصیات ایمونولوژیک بیماران با ITK deficiency شامل هیپوگاماگلوبولینمیای پیشرونده در کنار لنفوپنی و کاهش پیشرونده CD4+ Tcellها میباشد. همانند سایر افراد مبتلا به بیماری لنفوپرولیفراتیو وابسته به EBV، بیماران ITK deficient دارای سطوح شدیداً کاهش یافته NKT cell ها در خون محیطی هستند که نقش کلیدی در دفاع علیه ویروس EBV به عهده دارند (51). بر خلاف بیماران مبتلا به XLP1 که مستعد لنفوم بورکیت هستند این بیماران بیشتر با هوچکین مراجعه میکنند.
Autoimmune Lymphoprolifrative Syndrome (ALPS): سندرم لنفوپرولیفراتیو اتوایمیون یک اختلال ژنتیکی نادریست با زمینه ژنتیکی پیچیده که علاوه بر مشخصههای ژنتیکی مختلف الگوی توارثی آن نیز متفاوت میباشد. الگوهای توارثی اتوزومال غالب، اتوزومال مغلوب ومواردی موتاسیونهای سوماتیک نیز در این بیماران گزارش شده است. تظاهرات بالینی آن شامل: لنفادنوپاتی، ارگانومگالی، رویدادهای اتوایمیونیتی بهخصوص اتوایمیون سایتوپنی و لنفوم میباشد. هال مارک بیماری فنوتیپ ایمونولوژیک بهخصوصی است به صورت افزایشTCR α/β CD4-CD8- “double negative” T cellها. سایر بیومارکرهای مشاهده شده در ALPS شامل افزایش سطوح ویتامین B12، IL-10 ، soluble FAS ligand میباشد. در این بیماران اختلال در آپوپتوزیس وجود دارد که با واسطه FAS میباشد (53،52). علائم بالینی بیماران طیف وسیعی را شامل میشود که نتیجه هیپرپلازی غیر بدخیم لنفوییدی و تجمع پیشرونده سلولهای Auto reactive لنفوسیتهای T و B میباشد. میزان نفوذ علائم بالینی در بیماران بهطور کامل مشخص نشده است و بستگی به محل و نوع موتاسیون ژنتیکی دارد و حتی ممکن است در افرادی که موتاسیون مشابهی دارند علائم متفاوت باشد. اتوایمیون سایتوپنی شایعترین علامتی است که تمام ردههای خونی را میتواند درگیر کند. درگیری ارگانهای توپر شایع نیست و مواردیکه گزارش شده شامل گلومرولونفریت، هپاتیت، بافت همبند (لوپوس)، یووئیت، تیروئیدیت و گیلنباره بوده است. در مطالعهای که توسط Azizi و همکارانش روی 780 بیمار ALPS و ALPS-like syndrome صورت گرفت شایعترین تظاهر بالینی اسپلنومگالی (92/5%) و لنفادنوپاتی (86/8%) بود. اما در گروه ALPS-like بیماریهای هماتولوژیک (87/8%) و اتوایمیون (86/8%) شایعتر بود. در این بیماران بدخیمی به فرمهای متفاوتی وجود داشت. لنفوم هوچکین در 25 بیمار و لنفوم نانهوچکین در 26 بیمار گزارش شد. میزان بدخیمی بین دو گروه تفاوت معنیداری نداشت (54). بیماران باgerm line mutation در قسمت داخل سلولی پروتئین FAS ریسک بالاتری برای بروز لنفوم هوچکین و نان هوچکین دارند. در مطالعهای خطر نسبی لنفوم هوچکین و لنفوم نان هوچکین به ترتیب 51 و 14 بود. بیماران مبتلا به ALPS لنفادنوپاتی مزمن نوساندار دارند. بنابراین باید این افراد بهصورت مرتب جهت تشخیص زودهنگام لنفوم، توسط CT scan سریال و scan PET تحت مانیتورینگ قرار بگیرند (55).
نقایص سیستم ایمنی ذاتی
WHIM syndrome: سندرم WHIM یک نقص ایمنی نادری است که با حروف اختصاری کلمات Wart-hypogammaglobulinemia-Infection-myelokathexis تعریف میشود. میلوکاتکسی یک فرم غیر معمول از نوتروپنی مادرزادی غیرسیکلیک است که به علت تجمع نوتروفیلهای بالغ و دژنره در مغز استخوان ایجاد میشود. معمولاً مونوسیتوپنی و لنفوپنی بهخصوص B cell lymphopenia هم در این افراد دیده میشود. این بیماری توارث اتوزومال غالب دارد که ناشی از موتاسیون gain of function در ژن کدکننده G protein couple recetor(CXCR4) میباشد (56). اکثریت بیماران پان لکوسیتوپنی، نوتروپنی، لنفوپنی همراه با هیپوگاماگلوبولینمیا دارند. مطالعات، تنوع وسیعی را در فنوتیپ بالینی بیماران نشان داده است. انسیدانس متغیر در عفونتهای مکرر (عفونت تنفسی-عفونت پوستی - بیماری پریدونتال- استئومیلیت و مننژیت)، استعداد به عفونتهای HPV و EBV، نقایص کونوترانکال قلب و افزایش استعداد به بدخیمیها از جمله این علائم هستند. در کل تشخیص این بیماری بهصورت کلینیکوپاتولوژیکال است. اطلاعات کافی در مورد سیر طبیعی بیماری در بیماران وجود ندارد اما بعضی عوارض بیماری مثل برونشکتازی، کری و بدخیمیها را ممکن است با تشخیص زودرس بیماری پیشگیری کرد (57). این بیماران مستعد عفونت HPV هستند که باعث ایجاد زگیل در مخاط دهان-ناحیه تناسلی و پوست میشود و این حالت خود باعث افزایش ریسک اسکواموس کارسینوما میگردد. در یک مطالعه آنالیزی روی 18 بیمار مبتلا به سندرم WHIM صورت گرفت. انسیدانس زگیل در 61% بیماران و میزان بروز بدخیمیهای مرتبط با HPV 16 % گزارش شد (58). نوتروپنی مادرزادی (congenital neutropenia): نوتروپنی مادرزادی در برگیرنده گروهی از اختلالات است که با نوتروپنی پایدار یا متناوب شدید یا خفیف مشخص میشود. فیزیولوژیوپاتولوژی نوتروپنی در این بیماران هنوز بهطور دقیق مشخص نیست. ولی توقف تولید نوتروفیلها در مغز استخوان اثبات شده است. یک زنجیرهای بین نوتروپنی پایدار در مقابل نوتروپنی گذرا و موتاسیونها مختلف در لوکوس 3. 13 p 19 ژن کدکننده آنزیم نوتروفیل الاستاز (ELANE) وجود دارد. مکانیسمهای مولکولی که بهوسیله آن موتاسیونهای ELANE باعث اختلال در میلوپوئزیس (Myelopoiesis) میشود ناشناخته است. نوتروپنی مادرزادی شدید یک فرم نارسایی مغز استخوان است که بیمار از بدو تولد دارای نوتروپنی شدید همراه با استعداد به عفونتهای مختلف میباشد. نوتروپنی مرتبط با ELANE، سیکلیک نوتروپنی و نوتروپنی مادرزادی شدید (سندرم کاستمن) است که تقریباً فنوتیپ بالینی مشابهی دارند. موتاسیونهای EALNE در 100%-80% بیماران سیکلیک نوتروپنی و در 63%-35% موارد نوتروپنی مادرزادی شدید یافت میشود (59). بیماران مبتلا به severe congenital neutronpenia(SCN) دارای نوتروفیلهای خون محیطی کمتر از µ/200 هستند و اغلب همراه با سطوح افزایش یافته مونوسیتها میباشند. بیشترین موتاسیونی که در بیماران با ازدواجهای غیر فامیلی دیده میشود در ژن ELANE است در حالیکه میزان موتاسیون در ژن HAX1 در جمعیتهای با ازدواجهای فامیلی بیشتر دیده میشود. سابتایپهای SCN براسای الگوی توارثی شان و موتاسیون در ژنهای مختلف مشخص میشوند مثل موتاسیون در ژنهایSCF3R- JAGN1- VPS45- G6PC3- HAX1 که الگوی توارثی اتوزومال مغلوب دارند. ژنهای GFI1- ELANE- SRP54 توارث اتوزومال غالب و ژن WAS وابسته به X دارند (60). موتاسیونهای ELAN شایعترین علت SCN هستند که تقریباً 50% موارد را به خود اختصاص میدهند. در 30% موارد، علت ژنتیکی SCN ناشناخته است. علت مهم مورتالیتی در بیماران با SCN از زمان مصرف گسترده G-CSF، میلودیسپلازی و لوکمی میلوسیتیک حاد (AML/MDS) میباشد. رجیستری بیماران نوتروپنی در فرانسه، میزان بروز تجمعی AML یا MDS را 8/10% طی 20 سال در بیماران SCN گزارش کرده است. در مطالعه آیندهنگر دیگری روی 374 فرد مبتلا به SCN که به مدت طولانی تحت درمان G-CSF بودند بروز تجمعی AML/MDS بعد از 15 سال 22% اعلام شد. بیمارانی که به دوزهای بالاتر G-CSF نیاز داشتند و پاسخ ضعیفتری نشان میدادند در معرض ریسک بیشتری برای لوکمی بودند. لازم به ذکر است که AML وMDS در موتاسیونهایELAN- HAX1- G6PC3-WAS هم گزارش شده است (61).
نتیجهگیری
بیماران مبتلا به نقص سیستم ایمنی از آنجاییکه مستعد عفونتهای متعدد میباشند این قابلیت را دارند که بهدنبال برخی ویروسها و عوامل کارسینوژن دچار بدخیمیهای گوناگون بشوند (جدول 1). این افراد همچنین در بعضی موارد دارای اتوایمیونیتی و دیسرگولیشن ایمنی هستند که این خطرات را افزایش میدهد. از طرف دیگر وجود و تکرار بعضی بدخیمیها در خانوادهها به خصوص بدخیمیهای خونی این هشدار را به ما میدهد که ممکن است در پس این بدخیمیها، اختلالات سیستم ایمنی از جمله نقایص ایمنی اولیه نهفته باشد و لزوم ارزیابی سیستم ایمنی را در گروهی از این بیماران ایجاب میکند.
حامی مالی: ندارد
تعارض در منافع: وجود ندارد.
جدول 1 : بدخیمیهای هماتولوژیک و غیر هماتولوژیک در بیماران مبتلا به نقص ایمنی اولیه
References:
1- Khalili A. A Review of Primary Immunodeficiency Diseases with Skin Manifestations. J Shahid
Sadoughi Uni Med Sci 2022; 29(10): 4164-79.
2- Salavoura K, Kolialexi A, Tsangaris G, Mavrou A. Development of Cancer in Patients with Primary Immunodeficiencies. Anticancer Res 2008; 28(2B): 1263-9.
3- Rezaei N, Hedayat M, Aghamohammadi A, Nichols KE. Primary Immunodeficiency Diseases Associated with Increased Susceptibility to Viral Infections and Malignancies. J Allergy Clin Immunol 2011; 127(6): 1329-41.
4- Kebudi R, Kiykim A, Sahin MK. Primary Immunodeficiency and Cancer in Children; A Review of the Literature. Curr Pediatr Rev 2019; 15(4): 245-50.
5- Mayor PC, Eng KH, Singel KL, Abrams SI, Odunsi K, Moysich KB, et al. Cancer in Primary Immunodeficiency Diseases: Cancer Incidence in the United States Immune Deficiency Network Registry. J Allergy Clin Immunol 2018; 141(3): 1028-35.
6- Matza Porges S, Shamriz O. Genetics of Immune Dysregulation and Cancer Predisposition: Two Sides of the Same Coin. Clin Exp Immunol 2022; 210(2): 114-27.
7- Yazdani R, Habibi S, Sharifi L, Azizi G, Abolhassani H, Olbrich P، et al. Common Variable Immunodeficiency: Epidemiology، Pathogenesis، Clinical Manifestations، Diagnosis، Classification، and Management. J Investig Allergol Clin Immunol 2020; 30(1): 14-34.
8- Bruns L, Panagiota V, von Hardenberg S, Schmidt G, Adriawan IR, Sogka E, et al. Common Variable Immunodeficiency-Associated Cancers: The Role of Clinical Phenotypes، Immunological and Genetic Factors. Front Immunol 2022; 13: 742530.
9- Gullo I, Costa C, Silva SL, Ferreira C, Motta A, Silva SP, et al. The Dysfunctional Immune System in Common Variable Immunodeficiency Increases the Susceptibility to Gastric Cancer. Cells 2020; 9(6): 1498.
10- Kiaee F, Azizi G, Rafiemanesh H, Zainaldain H, Sadaat Rizvi F, Alizadeh M, et al. Malignancy in Common Variable Immunodeficiency: A Systematic Review and Meta-Analysis. Expert Rev Clin Immunol 2019; 15(10): 1105-13.
11- Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin 2018; 68(6): 394-424.
12- Leone P, Vacca A, Dammacco F, Racanelli V. Common Variable Immunodeficiency and Gastric Malignancies. Int J Mol Sci 2018; 19(2): 451.
13- Pulvirenti F, Pecoraro A, Cinetto F, Milito C, Valente M, Santangeli E, et al. Gastric Cancer is the Leading Cause of Death in Italian Adult Patients with Common Variable Immunodeficiency. Front Immunol 2018; 9: 2546.
14- Tak Manesh A, Azizi G, Heydari A, Kiaee F, Shaghaghi M, Hossein-Khannazer N, et al. Epidemiology and Pathophysiology of Malignancy in Common Variable Immunodeficiency? Allergol Immunopathol (Madr) 2017; 45(6): 602-15.
15- Khalili A, Yadegari AH, Delavari S, Yazdani R, Abolhassani H. Disseminated Intravascular Coagulation Associated with Large Deletion of Immunoglobulin Heavy Chain. Iran J Allergy Asthma Immunol 2021; 20(6): 778-83.
16- Khalili A, Plebani A, Vitali M, Abolhassani H, Lougaris V, Mirminachi B, et al. Autosomal Recessive Agammaglobulinemia: A Novel Non-Sense Mutation in CD79a. J Clin Immunol 2014; 34(2): 138-41.
17- Bagheri Y, Vosughi A, Azizi G, Yazdani R, Kiaee F, Hafezi N, et al. Comparison of Clinical and Immunological Features and Mortality in Common Variable Immunodeficiency and Agammaglobulinemia Patients. Immunol Lett 2019; 210: 55-62.
18- Hajjar J, Hasan S, Forbes LR, Hemmige V, Orange JS. Gastric Adenocarcinoma in a Patient with X-Linked Agammaglobulinemia and HIV: Case Report and Review of the Literature. Front Pediatr 2016; 4: 100.
19- Brosens LA, Tytgat KM, Morsink FH, Sinke RJ, Ten Berge IJ, Giardiello FM, et al. Multiple Colorectal Neoplasms in X-Linked Agammaglobulinemia. Clinical Gastroenterology and Hepatology 2008; 6(1): 115-9.
20- Gokce G, Ceylan OM, Uysal Y, Yildizoglu U, Atas E, Kurt B. Epiphora as the Presenting Sign of Relapsed Non-Hodgkin Lymphoma in a Child with Bruton Agammaglobulinemia. Eur J Ophthalmol 2015; 25(1): 65-7.
21- Moeini Shad T, Yazdani R, Amirifar P, Delavari S, Heidarzadeh Arani M, Mahdaviani SA, et al. Atypical Ataxia Presentation in Variant Ataxia Telangiectasia: Iranian Case-Series and Review of the Literature. Front Immunol 2022; 12: 779502.
22- Mitiagin Y, Barzilai A. Ataxia-Telangiectasia Mutated Plays an Important Role in Cerebellar Integrity and Functionality. Neural Regen Res 2023; 18(3): 497-502.
23- Bakhtiar S, Salzmann-Manrique E, Donath H, Woelke S, Duecker RP, Fritzemeyer S, et al. The Incidence and Type of Cancer in Patients with Ataxia‐Telangiectasia Via a Retrospective Single‐Centre Study. Br J Haematol 2021; 194(5): 879-87.
24- Oska S, Zarbo A, Yeager D, Friedman BJ, Shwayder T. Melanoma Arising in a Patient with Ataxia-Telangiectasia: A Call for Full Skin Examinations in This Patient Population. Pediatr Dermatol 2020; 37(4): 767-8.
25- Hosahalli Vasanna S, Pereda MA, Dalal J. Clinical Features، Cancer Biology، Transplant Approach and other Integrated Management Strategies for Wiskott–Aldrich Syndrome. J Multidiscip Healthc 2021; 14: 3497-512.
26- Sullivan KE, Mullen CA, Blaese RM, Winkelstein JA. A Multiinstitutional Survey of the Wiskott-Aldrich Syndrome. J Pediatr 1994; 125(6 Pt 1): 876-85.
27- Menotti M, Ambrogio C, Cheong TC, Pighi C, Mota I, Cassel SH, et al. Wiskott–Aldrich Syndrome Protein (WASP) is a Tumor Suppressor in T Cell Lymphoma. Nat Med 2019; 25(1): 130-40.
28- Perry GS 3rd, Spector BD, Schuman LM, Mandel JS, Anderson VE, McHugh RB, et al. The Wiskott-Aldrich Syndrome in the United States and Canada (1892–1979). J Pediatr 1980; 97(1): 72-8.
29- Cekic S, Metin A, Aytekin C, Edeer Karaca N, Baris S, Karali Y, et al. The Evaluation of Malignancies in Turkish Primary Immunodeficiency Patients; A Multicenter Study. Pediatr Allergy Immunol 2020; 31(5): 528-36.
30- Tsilifis C, Freeman AF, Gennery AR. STAT3 Hyper-Ige Syndrome—An Update and Unanswered Questions. J Clin Immunol 2021; 41(5): 864-80.
31- Haskologlu S, Kostel Bal S, Islamoglu C, Aytekin C, Guner S, Sevinc S, et al. Clinical، Immunological Features and Follow Up of 20 Patients with Dedicator of Cytokinesis 8 (DOCK8) Deficiency. Pediatr Allergy Immunol 2020; 31(5): 515-27.
32- Aydin SE, Kilic SS, Aytekin C, Kumar A, Porras O, Kainulainen L, et al. DOCK8 Deficiency: Clinical and Immunological Phenotype and Treatment Options-A Review of 136 Patients. J Clin Immunol 2015; 35(2): 189-98.
33- Sharapova SO, Pashchenko OE, Bondarenko AV, Vakhlyarskaya SS, Prokofjeva T, Fedorova AS, et al. Geographical Distribution، Incidence، Malignancies، and Outcome of 136 Eastern Slavic Patients with Nijmegen Breakage Syndrome and NBN Founder Variant C. 657_661del5. Front Immunol 2021; 11: 602482.
34- El Hasbaoui B، Elyajouri A، Abilkassem R، Agadr A. Nijmegen Breakage Syndrome: Case Report and Review of Literature. Pan Afr Med J 2020; 35: 85.
35- Sharma R, Lewis S, Wlodarski MW. DNA Repair Syndromes and Cancer: Insights into Genetics and Phenotype Patterns. Front Pediatr 2020; 8: 570084.
36- Izawa K, Martin E, Soudais C, Bruneau J, Boutboul D, Rodriguez R, et al. Inherited CD70 Deficiency in Humans Reveals a Critical Role for the CD70–CD27 Pathway in Immunity to Epstein-Barr Virus Infection. J Exp Med 2017; 214(1): 73-89.
37- Abolhassani H, Edwards ES, Ikinciogullari A, Jing H, Borte S, Buggert M, et al. Combined Immunodeficiency and Epstein-Barr Virus–Induced b Cell Malignancy in Humans with Inherited CD70 Deficiency. J Exp Med 2017; 214(1): 91-106.
38- Abolhassani H. Specific Immune Response and Cytokine Production in CD70 Deficiency. Frontiers in Pediatrics 2021; 9: 615724.
39- Han BK, Olsen NJ, Bottaro A. The CD27–CD70 Pathway and Pathogenesis of Autoimmune Disease. Semin Arthritis Rheum 2016: 45(4): 496-501.
40- Ghosh S, Köstel Bal S, Edwards ESJ, Pillay B, Jiménez Heredia R, Erol Cipe F, et al. Extended Clinical and Immunological Phenotype and Transplant Outcome in CD27 and CD70 Deficiency. Blood 2020; 136(23): 2638-55.
41- Lino CNR, Ghosh S. Epstein–Barr Virus in Inborn Immunodeficiency—More than Infection. Cancers (Basel) 2021; 13(19): 4752.
42- Xu T, Zhao Q, Li W, Chen X, Xue X, Chen Z, et al. X-Linked Lymphoproliferative Syndrome in Mainland China: Review of Clinical، Genetic، and Immunological Characteristic. Eur J Pediatr 2020; 179(2): 327-38.
43- El-Mallawany NK, Curry CV, Allen CE. Haemophagocytic Lymphohistiocytosis and Epstein–Barr Virus: A Complex Relationship with Diverse Origins، Expression and Outcomes. Br J Haematol 2022; 196(1): 31-44.
44- Szmyd B, Mlynarski W, Pastorczak A. Genetic Predisposition to Lymphomas: Overview of Rare Syndromes and Inherited Familial Variants. Mutat Res Rev Mutat Res 2021; 788: 108386.
45- Liang JH, Zhu HY, Xu DM, Wang L, Wang Y, Qiao C, et al. A New SH2D1A Mutation in a Female Adult XLP Disease with Hemophagocytic Lymphohistiocytosis and NK-Cell Leukemia. Ann Hematol 2019; 98(12): 2829-31.
46- Abbas R, Larisch S. Targeting XIAP for Promoting Cancer Cell Death—The Story of ARTS and SMAC. Cells 2020; 9(3): 663.
47- Mudde ACA, Booth C, Marsh RA. Evolution of Our Understanding of XIAP Deficiency. Front Pediatr 2021; 9: 660520.
48- Latour S, Winter S. Inherited Immunodeficiencies with High Predisposition to Epstein–Barr Virus-Driven Lymphoproliferative Diseases. Front Immunol 2018; 9: 1103.
49- Duan L, Grunebaum E. Hematological Malignancies Associated with Primary Immunodeficiency Disorders. Clinical Immunology 2018; 194: 46-59.
50- Wallace JG, Alosaimi MF, Khayat CD, Jaber F, Almutairi A, Beaussant-Cohen S, et al. ITK Deficiency Presenting as Autoimmune Lymphoproliferative Syndrome. J Allergy Clin Immunol 2021; 147(2): 743-5. e1.
51- Ghosh S, Bienemann K, Boztug K, Borkhardt A. Interleukin-2-Inducible T-Cell Kinase (ITK) Deficiency-Clinical and Molecular Aspects. J Clin Immunol 2014; 34(8): 892-9.
52- Consonni F, Gambineri E, Favre C. ALPS، FAS، and Beyond: From Inborn Errors of Immunity to Acquired Immunodeficiencies. Ann Hematol 2022; 101(3): 469-84.
53- Li P, Huang P, Yang Y, Hao M, Peng H, Li F. Updated Understanding of Autoimmune Lymphoproliferative Syndrome (ALPS). Clin Rev Allergy Immunol 2016; 50(1): 55-63.
54- Hafezi N, Zaki-Dizaji M, Nirouei M, Asadi G, Sharifinejad N, Jamee M, et al. Clinical، Immunological، and Genetic Features in 780 Patients with Autoimmune Lymphoproliferative Syndrome (ALPS) and ALPS‐Like Diseases: A Systematic Review. Pediatr Allergy Immunol 2021; 32(7): 1519-32.
55- Leechawengwongs E, Shearer WT. Lymphoma Complicating Primary Immunodeficiency Syndromes. Curr Opin Hematol 2012; 19(4): 305-12.
56- Heusinkveld LE, Majumdar S, Gao JL, McDermott DH, Murphy PM. WHIM Syndrome: From Pathogenesis Towards Personalized Medicine and Cure. Journal of Clinical Immunology 2019; 39(6): 532-56.
57- Geier CB, Ellison M, Cruz R, Pawar S, Leiss-Piller A, Zmajkovicova K, et al. Disease Progression of WHIM Syndrome in an International Cohort of 66 Pediatric and Adult Patients. J Clin Immunol 2022; 42(8): 1748-65.
58- Tiri A, Masetti R, Conti F, Tignanelli A, Turrini E, Bertolini P, et al. Inborn Errors of Immunity and Cancer. Biology (Basel) 2021; 10(4): 313.
59- Lazzareschi I, Rossi E, Curatola A, Capozio G, Benacquista L, Iezzi L, et al. Assessment of Congenital Neutropenia in Children: Common Clinical Sceneries and Clues for Management. Mediterr J Hematol Infect Dis 2022; 14(1): e2022008.
60- Hojabri M, Farsi Y, Jamee M, Abolhassani H, Khani HHK, Karimi A, et al. JAGN1 Mutation with Distinct Clinical Features; Two Case Reports and Literature Review. BMC Pediatr 2023; 23(1): 206.
61- Link DC. Mechanisms of Leukemic Transformation in Congenital Neutropenia. Curr Opin Hematol 2019; 26(1): 34-40.
نوع مطالعه: مروری |
موضوع مقاله:
ایمونولوژی
دریافت: 1402/2/31 | پذیرش: 1402/7/9 | انتشار: 1402/11/15
دریافت: 1402/2/31 | پذیرش: 1402/7/9 | انتشار: 1402/11/15
ارسال پیام به نویسنده مسئول
| بازنشر اطلاعات | |
 |
این مقاله تحت شرایط Creative Commons Attribution-NonCommercial 4.0 International License قابل بازنشر است. |
