

دوره 30، شماره 11 - ( بهمن 1401 )
جلد 30 شماره 11 صفحات 6076-6052 |
برگشت به فهرست نسخه ها


![]()
![]()
![]()
Download citation:
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:



BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
Khatami M, Heidari M M, Mazrouei B, Aflaki R. Review of Genetic Changes and Factors Causing Cardiac Arrhythmias. JSSU 2023; 30 (11) :6052-6076
URL: http://jssu.ssu.ac.ir/article-1-5805-fa.html
URL: http://jssu.ssu.ac.ir/article-1-5805-fa.html
خاتمی مهری، حیدری محمد مهدی، مزروعی بهاره، افلاکی رژین. مروری بر تغییرات ژنتیکی و عوامل ایجادکننده آریتمیهای قلبی. مجله علمي پژوهشي دانشگاه علوم پزشكي شهید صدوقی يزد. 1401; 30 (11) :6052-6076



متن کامل [PDF 1737 kb]
(475 دریافت)
| چکیده (HTML) (1100 مشاهده)
متن کامل: (2716 مشاهده)
مقدمه
قلب یک عضو عضلانی است که بهطور مداوم، یعنی در حدود 3 بیلیون سیکل در طول حیات یک فرد، خون را به تمام بدن پمپ میکند و دارای دو حفره دهلیزی و دو حفره بطنی میباشد. یک ضربان قلب ساده از انبساط، یعنی زمانی که دهلیزها و بطنها از خون پر شده و انقباض، زمانیکه خون به کل بدن پمپ میشود، تشکیل شده است (1,2). آریتمی به معنی ریتم غیرطبیعی ضربان قلب است، انواع مختلفی از آریتمی وجود دارد که موجب ایجاد ضربان خیلی سریع (تاکیکاردی) یا خیلی آهسته (برادیکاردی) میشود و درنتیجه قلب پمپاژ غیر مؤثری را انجام میدهد. در واقع در اثر اختلال سیستم هدایت الکتریکی طبیعی قلب، بیماریهای آریتمی قلبی ایجاد میشوند (شکل1). آریتمیها شایع هستند و میلیونها نفر در جهان را درگیر کردهاند و به عنوان یکی از علل اصلی مرگهای ناگهانی در آمریکا شناخته شدهاند که سالیانه موجب مرگ ۴۰۰۰۰۰ نفر میشوند. فیبریلاسیون دهلیزی شایعترین شکل آریتمی در افراد مسن در آمریکاست که تقریباً 2/5 میلیون نفر به آن مبتلا هستند (3،4).
امواج پلاریزه و دپلاریزه: انقباضات منظم قلب وابسته به یک شبکه الکتریکی است که امواج الکتریکی را به تمام قلب هدایت میکند. ضربانساز غالب قلب، گره سینوسی دهلیزی، آغازگر موج دپولاریزاسیونی است که به شکل یک موج گسترش یافته و دهلیزها را برای انقباض تحریک میکند. این گره سینوسی در داخل دیواره خلفی فوقانی دهلیز راست قرار دارد و بهطور طبیعی، پالس الکتریکی ایجاد شده از طریق رشتههای عضلانی به تمام قلب توزیع میشود. داخل سلولهای قلب، در حال استراحت، بار منفی وجود دارد (سلولها پولاریزهاند)، اما زمانی که با تحریک الکتریکی دپولاریزه میشوند، منقبض میشوند. موج دپولاریزاسیون (مثبت شدن بار داخل سلولها) و مرحله رپولاریزاسیون (برگشت بار منفی داخل سلولها) متعاقب آن، روی اکوکاردیوگرام (ECG) ثبت میگردد (شکل2) (5). تحریک الکتریکی دپولاریزاسیون در داخل دهلیزها منتشر میشود و باعث ایجاد موج P بر روی الکتروکاردیوگرام میشود که نشاندهنده انقباض همزمان دهلیزهاست. تحریک الکتریکی از طریق تارهای انتهایی فیبرهای پورکنژ، دپولاریزاسیون را به سلولهای میوکارد بطنی میرساند. دپولاریزاسیون میوکارد بطنی باعث ایجاد کمپلکس QRS بر روی ECG و انقباض بطنها میشود. پس از هر کمپلکس QRS، یک خط ایزوالکتریک افقی دیده میشود که قطعه ST نامیده میشود و پس از آن یک موج T عریض ظاهر میشود. موج T، نشاندهنده فاز سریع انتهایی رپولاریزاسیون بطنی است که در طی آن رپولاریزاسیون با سرعت و بهطور موثر روی میدهد. از آنجا که سیستول یا انقباض بطنی از آغاز QRS تا پایان موج T به طول میانجامد، فاصله QT (QT interval) از لحاظ بالینی اهمیت زیادی دارد (شکل 3). فاصله QT (QT interval) در اکوکاردیوگرام، نشاندهنده دوره فعالیت و بازگشت میوکاردیوم بطنی است. در واقع فاصله QT، زمانی است که سپری میشود تا بطنها دپولاریزه (شروع QRS) و رپولاریزه (انتهای موج T) شوند و این مدت بسته به میزان تپش قلب دارد (6،7).
عامل ایجاد ایمپالسهای الکتریکی قلب: ایمپالس الکتریکی توسط گرادیانت الکتروشیمیایی موجود در غشاء سلولهای عضله قلبی یا کاردیومیوسیتها ایجاد میشود و به تعادل جریان انتقال یونها در داخل و خارج غشاء سلول وابسته است (8). در حفظ این تعادل، کانالهای یونی از اهمیت خاصی برخوردارند و میتوان گفت، اساس مولکولی الکتروفیزیولوژی قلب، کانالهای یونی هستند (3). در ژنوم انسان، حدود 429 کانال یونی وجود دارد و تقریبا 30 ژن کدکننده کانالهای یونی قلب تاکنون شناسایی شدهاند (9,10).
کانالهای یونی: کانالهای یونی قلب، حاوی پروتئینها و گلیکوپروتئینهایی هستند که در سارکولمای کاردیومیوسیتها واقعاند و تشکیل منافذی در غشاء سلول را میدهند که به یونهای خاصی اجازه میدهند، بر اساس شیب الکتروشیمیایی از غشاء عبور کنند که به این ترتیب باعث تنظیم عملکرد سلول میشوند (8,10). چهار نوع کلی از این کانالها وجود دارد: کانالهای بدون دروازه یعنی همواره باز (Non-gatedchannels): مانند پمپهای سدیم و پتاسیم، کانالهای دارای دروازه (Directly gated channels): مانند کانالهای وابسته به ولتاژ (Voltage-gated channels) و وابسته به لیگاند (Ligand-gated channels)، کانالهای وابسته به پیامبرهای ثانویه (Second messenger gated channels): مانند گیرنده¬های پروتئین G (G-protein receptors) و کانالهای وابسته به ذخیره (Store-operated channels): مانند کانالهای پتانسیل موقتی رسپتور (Transientreceptor potential channels) (11). کانالهای یونی برای طیف وسیعی از عملکردهای فیزیولوژیکی مانند سیگنالهای نورونی، انقباضات عضلانی، هدایت عمل قلب، ترشح هورمونی، تنظیم حجم سلول و تکثیر سلولی ضروری هستند و به همین دلیل کانالهای یونی در بیماریهای زیادی دخالت دارند که اغلب آنها بیماریهای توارثی هستند و در نتیجه جهشهایی در ژنهای کدکننده پروتئینهای کانال ایجاد میشوند (11,12). کانالهای وابسته به ولتاژ عصب و عضله از نظر گسترش ایمپالسهای عصبی و انقباضات عضلانی حائز اهمیت هستند. بهطور مثال، کانالهای سدیم با دپولاریزاسیون غشا سلول، فعال میشوند. در حالت باز، آنها بهطور انتخابی اجازه ورود یونهای سدیم را میدهند. جریان یونها به داخل سلول، تولید دپولاریزاسیون موضعی قوی به نام پتانسیل عمل میکند که باعث باز شدن کانالهای سدیم وابسته به ولتاژ جدیدی میگردد که دپولاریزاسیون را شدت میدهد. پس میتوان گفت کانالهای یونی وابسته به ولتاژ، زمینه ساز پتانسیل عمل در سلولهای عضله قلبی هستند. نقص در هر کدام از این کانالهای یونی منجر به اختلال در پتانسیل عمل سلولهای عضله قلبی و اختلالات اکوکاردیوگرام و باعث ایجاد زمینهای برای بروز آریتمیهای قلبی میشود (13).
بیماریهای قلب: بهطور کلی دو نوع بیماری مهم در قلب وجود دارد:
1) کاردیومیوپاتی که در اثر تغییراتی در پروتئینهای سارکومریک و اسکلتسلولی رخ میدهد.
2) بیماری¬های آریتموژنیک که توسط جهشهایی در کانالهای یونی و پروتئینهای کنترل کننده کانال ایجاد میشوند که به این گونه بیماریها،Cardiac channelopathies میگویند. نظیر: سندرم¬های Long QT (LQTs) و بروگادا (Brugada syndrome) و Short QT (SQTs)، تاکی کاردیای دهلیزی (CatecholaminergicPolymorphic Ventricular Tachycardias) و فیبریلاسیون ایدیوپاتیک (13،14).
آریتمیهای قلبی: بیماریهای آریتموژنیک قلب، عوامل مهم مرگهای ناگهانی قلبی در افراد جوان با قلبهای سالم هستند (6). در سالهای اخیر مطالعات گستردهای روی ژنهای کانالهای یونی قلب انجام شده است، اما هنوز 30 تا 45% موارد آریتمی را نمیتوان با این جهشهای شناخته شده توضیح داد (13,15). آریتمی یا ضربان غیر طبیعی قلب، ممکن است بهصورتتغییر در سرعت و یا نظم ضربانهای قلب باشد. در جریان آریتمی، ضربان ممکن است بیش از حد آهسته، بسیار تند و یا نامنظم باشد. آریتمی ممکن است به شکلهای مختلف تظاهر کند. میتواند بهصورت احساس لرزش یا ریزش در سینه (طپش قلب) همراه با درد سینه باشد. گاه باعث سبکی در سر میشود و یا بهصورت حملاتی همراه با بیهوش شدن است. در مواردی هم آریتمی، علامت مهمی ندارد و بیمار به آن توجهی نمیکند، اما وقتی ضربان قلب به حدی کند یا تند باشد که در عملکرد قلب به عنوان یک پمپ، اختلال ایجاد کند، میتواند بیمار را با خطری جدی مواجه سازد (13,15). تعیین ژنهایی که باعث بروز سندرمهای آریتموژنیک توارثی میشوند، مبنای مطالعات مولکولی است که روش تشخیصی جدیدی را علاوه بر اکوکاردیوگرام در اختیار محققین قرار میدهد (شکل5).
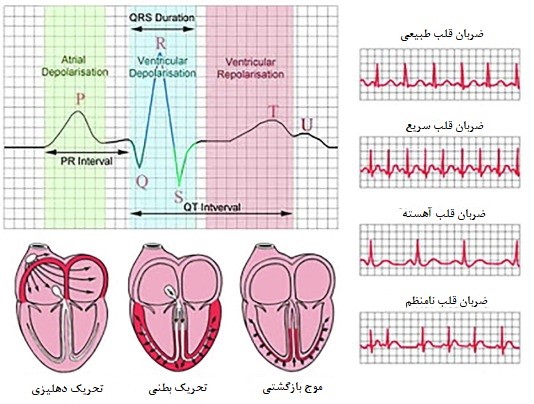
شکل1: مقایسه امواج الکتریکی قلب نرمال و قلب دارای آریتمی (https://www.ashwinihospital.co/ECG.html)
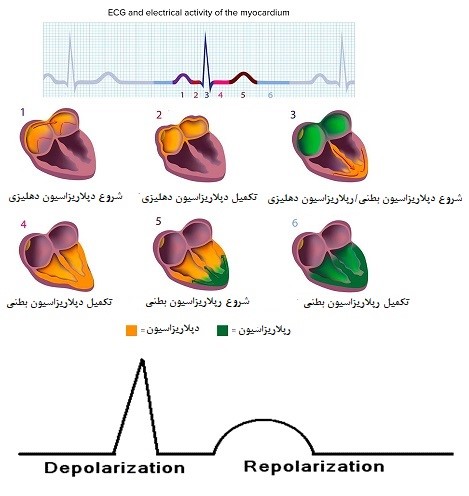
شکل2: دو موج دپلاریزاسیون ( انقباض) و رپلاریزاسیون( استراحت) را در ECG نشان میدهد (5)
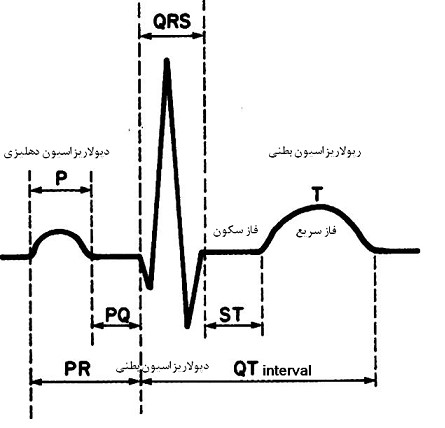
شکل3: اندازه گیری فاصلهQT در اکوکاردیوگرام (ECG). دیاگرام نشاندهنده ECG نرمال با موج P ( فعالیت دهلیزی)، مجموعه QRS (فعالیت بطنی و شروع انقباض بطنی) و موج T (رپلاریزاسیون بطنی) است. فاصله QT به طول بین شروع موج Q تا انتهای موج T گفته میشود
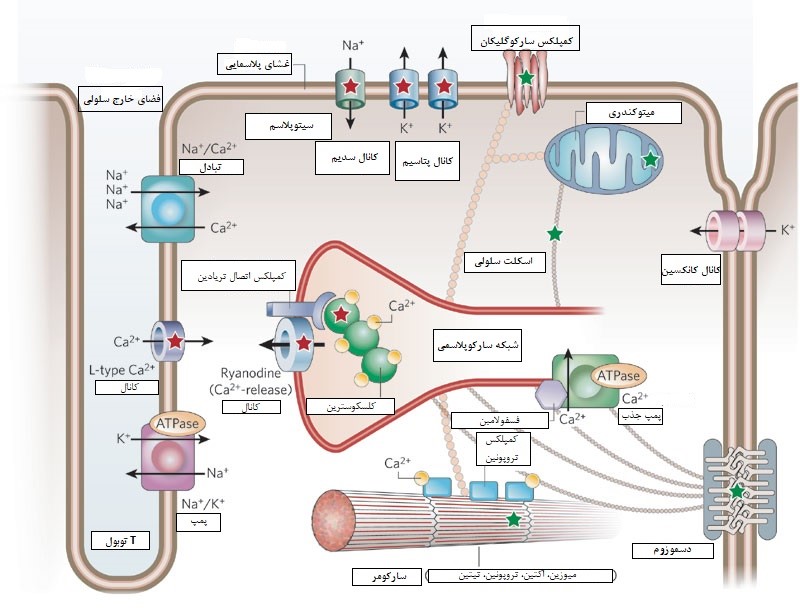
شکل4: ساختار کاردیومیوسیت بطنی (11)

شکل 5: تاثیر جهش ژنهای مختلف دخیل در بیماریهای قلبی در منحنی اکوکاردیوگرام (4)
نقصهای متابولیکی در بیماران آریتمی: مشخص شده است که متابولیسم میوکاردیال (Myocardial metabolism) میتواند خود را با تعداد زیادی از سوبستراها وفق دهد، اما الگوی دقیق مصرف سوبسترا بر اساس میزان دسترس بودن، تحویل اکسیژن، حجم و تنظیم فیزیولوژی آنها است. بر این اساس اغلب نقصهای متابولیکی تک ژنی می-توانند باعث اختلال در عملکرد کاردیومیوسیتها و در نهایت آریتمی و مرگ ناگهانی فرد شوند (14,15). چون فعالیت بسیاری از کانالهای یونی وابسته به متابولیسم سلولی است، بنابراین نقصهای ژنوم میتوکندری، ممکن است اثرات متفاوتی در بروز بیماری داشته باشند، مانند تفاوتهای هتروپلاسمی در بافتهای مختلف. از میان این بافتها، قلب بیشترین آسیبپذیری را نسبت به جهشهای میتوکندری دارد که غالباً بهصورت اختلالات قلبی بروز پیدا میکند (16). این امر به دلیل ارتباط بسیار نزدیک نقصهای متابولیسم و آریتمیهای قلبی است که ممکن است در سنین بلوغ بیشتر دیده شوند (14,15). دلایل متعددی وجود دارد که جهشهای ژنوم میتوکندری ممکن است در سندرمهای مرگ ناگهانی (Sudden death) دخالت داشته باشند که از آن جمله می-توان به فرکانس، شیوع نسبتا بالا و ارتباط جنسیت با وقوع بیماری اشاره کرد که نشاندهنده عدم توارث مندلی در این بیماران است (17). احتمال بالایی وجود دارد که الگوی توارثی بهصورت پلیژنیک بوده و هر دو ژنوم هستهای و میتوکندریایی در بروز آن دخیل باشند (14). اگر چه بروز بیماری در مردان بیشتر از زنان دیده شده است، اما این امر نمیتواند به دلیل توارث وابسته به X باشد، چرا که در خانوادههای درگیر، مادران بیشتر از پدران، علایم بیماری را نشان می¬دهند و این فرضیه با قدرت مطرح میشود که ژنوم میتوکندری و جهشهای آن ممکن است در روند آریتمی دخالت داشته باشند (14).
انواع آریتمی:
1. سندرم Long QT (LQTS)
سندرم Long QT نوعی بیماری رپولاریزاسیون میوکاردیال است. این آریتمی بدخیم ناشی از کانالوپاتی یونی قلب است که منجر به تاخیر در دو قطبی شدن پتانسیل عمل قلب میشود و با طویل شدن فاصله QT روی اکوکاردیوگرام، قابل تشخیص است (19,18). این سندرم در واقع نوعی آریتموژنیک بطنی (Ventricular arrhythmogenic) است که میتواند بهصورت توارثی یا اکتسابی باشد. بیماران LQTs مستعد برای بیماری Torsade de Pointes بطنی (TdP) هستند و بهطور جدی در معرض مرگهای ناگهانی قلبی (SCD)حتی در صورت داشتن قلب به ظاهر سالم میباشند (17). علائم این سندرم در بیماران، شامل حمله قلبی، طپش قلب در طی ورزش یا هیجانات زیاد و سنکوپ میباشد و در شرایطی هم اولین نشانه، ایست قلبی (Cardiac arrest) است. در موارد اکتسابی، مهمترین دلیل بروز LQTs استفاده از برخی داروهای قلبی است. سه اختلال سندرم انکرین-B (Ankyrin-Bsyndrome)، اندرسون تاویل (Andersen-Tawil syndrome) و تیموتی (Timothy syndrome) هم باعث LQT میشوند (17,20). استرس و اضطراب هم در بروز LQTs اکتسابی بیتاثیر نیست و تحقیقات نشان داده که افراد مبتلا به سندرم، موقعیتهای استرسزای بیشتری را تجربه کرده بودند (21). در LQTs اکتسابی هم، اختلال در مکانیسمهای یونی، مشابه با نوع توارثی آن دیده میشود (17). تاریخچه و انواع LQTS: گزارشهایی در مورد بروز سندرم LQT وجود دارد که فرزندان در خانوادههایی دچار مرگ ناگهانی قلبی شده بودندکه بعضاً یا طی ورزش دچار سنکوپ شده بودند یا استرس و فعالیتهای هیجانی را در سن های 4، 5، 8 و 9 سالگی تجربه کرده بودند (17). طویل شدن فاصله QT در ECG آنها کاملاً مشخص بود و توارث بیماری در آنها اتوزومال مغلوب شناخته شد. سندرم مشابه دیگری با علایم مرگ ناگهانی طی ورزش یا استرسهای هیجانی اما با توارث اتوزومال غالب، در مطالعات بعدی گزارش شد. این دو فرم از LQTs توارثی، اکنون به عنوان سندرمهای ژرویل، لانگ- نیلسن (Jervell، Lange-Nielsen syndrome) و رومانو-وارد (Romano-Ward syndrome) شناخته میشوند (19). دو سندرم دیگر هم طی سالهای اخیر شناسایی شدهاند به نام های آندرسن تاویل و سندرم تیموتی که گاه آنها را به عنوان LQT7 نام میبرند (18). میزان نفوذ (Prevalence) LQTs توارثی در آمریکا، حدود 1 فرد از 7000 فرد تخمین زده شده است و شاید هر ساله باعث مرگ ناگهانی 3000 -2000 نفر کودک و جوان شود. نوع رومانو-وارد، حدود 99% موارد (با میزان نفوذ 1:5000 تا 1:10000) را شامل میشود و فرم ژرویل، لانگنیلسن نادرتر است و در کمتر از 1% بیماران گزارش شدهاست (با میزان نفوذ 1:55000 تا 1:200000) (17).
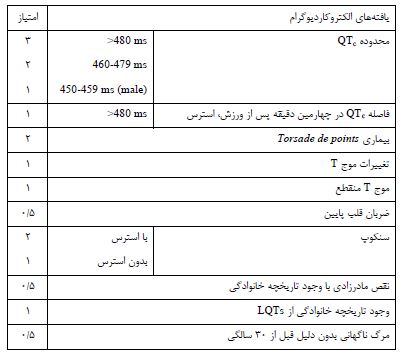
ژنتیک LQTS: مطالعات مولکولی ثابت کرده است کانالهای یونی که فعالیت الکتریکی قلب را کنترل میکنند، در بروزLQTs های توارثی نقش دارند و در این ارتباط، جهشهایی در ژنهای کدکننده کانالهای یونی قلب شناسایی شده است (22). این ژنها را بر اساس ترتیب کشف، نامگذاری می-کنند مانند: LQT1 ،LQT2 و ... این سندرم از نظر ژنتیکی، بیماری هتروژنوسی است که با جهشهای شناخته شدهای در ژنهای کدکننده کانالهای یونی قلب، همراه است که تعدادی از آنها کانال پتاسیم را کد میکنند و یکی کدکننده کانال سدیم (SCN5A) است. محققان تاکنون بیش از 200 جهش را در بیماران LQT یافتهاند. اما در اغلب خانوادهها، جهشهای جدیدی را هم میتوان یافت (23). جدول 1، ژنها، پروتئینهای کد شده و کانالهای یونی مربوطه را نشان میدهد (20-18،14).
اساس ژنتیکی :QTs17 زیرگونه مختلف LQTs وجود دارد که با جهشهای تک ژنی 15 ژن غالب اتوزومال مرتبط هستند (18). در حال حاضر سه ژن اصلی KCNQ1 وKCNH2 و SCN5A وجود دارد که تقریباً 75 درصد این اختلال را دربرمیگیرند و سایر ژنهای کشف شده، در مجموع کمتر از 5 درصد موارد LQTs را تشکیل میدهند (شکل6) (25-24،20). LQTs بر اساس جهشهای مرتبط با 15 ژن غالب اتوزومال، LQT 1-15، به 17 زیرگروه طبقهبندی میشوند. از این رو، جهش در ژنهای KCNQ1، HERG ، SCN5A ، KCNE1، KCNE2 و KCNJ2 به ترتیب باعث شکل LQT1، LQT2، LQT3، LQT5، LQT6 و LQT7 از LQTS میشود. که سه تا از مهمترین آنها را در ادامه بررسی میکنیم (شکل7) (18,20). LQT1: یکی از زیرگروههای شایع این بیماری است. بیماران مبتلا به این نوع LQT1، که حدود 42 درصد از کل بیماران مبتلا به LQTSهای مادرزادی را تشکیل میدهند، معمولاً قبل از 10 سالگی مراجعه میکنند (26). دلیل اصلی ایجاد این بیماری، اختلال در عملکرد ژن KCNQ1 است که کدکننده زیر واحد α کانال پتاسیم دارای ولتاژ (KV7.1) موجود در غشای سلولی کاردیومیوسیتها است. در واقع KV7.1 موتانت، جریان پتاسیم تأخیری را به آرامی فعال میکند (18,20) و هنگامی که با زیر واحدهای نرمال دیگر ترکیب میشود، پروتئینهای کانالی ناکارآمدی را تشکیل میدهند که به طور غیرطبیعی چین خوردهاند و معمولاً تحت تجزیه سریع قرار میگیرند. کانال KV7.1 از چهار زیر واحد α تشکیل شده است که برای برقراری جریان پتاسیم، با زیرواحدهای β ژن KCNE ترکیب میشوند. زیر واحد α ژن KCNQ1 دارای یک دامین سنجش ولتاژ (S1-4)، یک دامین تشکیل منفذ (S5-6) و همچنین دامینهای انتهای N و C درون سلولی است. LQT1 بر روی الکتروکاردیوگرام سطحی به عنوان یک موج T گسترده و متقارن با یک وقفه QTc طولانی مدت ظاهر میشود (21). لازم به ذکر است که ورزش جسمانی و تحریک سمپاتیک باعث تحریک سنکوپ و مرگ ناگهانی در بیماران مبتلا به LQT1 میشود (شکل8)(24).
جدول 1: ژنهای هستهای ایجاد کننده LQTs. فلش رو به بالا (↑) یا پایین (↓) به ترتیب افزایش یا از دست دادن عملکرد پروتئین را نشان میدهد (24،20-18).
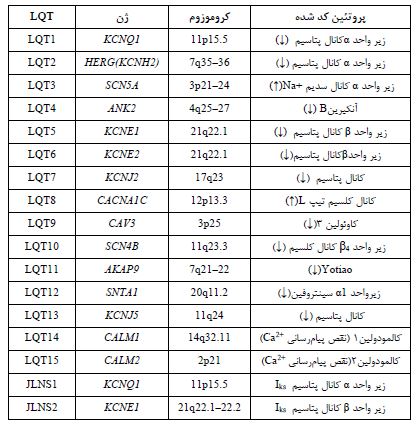

شکل6: خلاصه ژنهای مستعد سندرم QT طولانی (25)
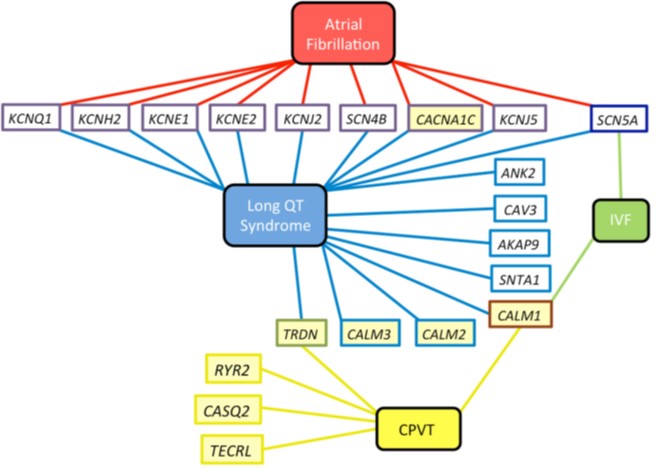
شکل 7: ژنهای دخیل در اختلالات آریتمی ژنتیکی (18)

شکل8: نقص کانال یونی در بروز LQT1 و اثر آن بر الکتروکاردیوگرام (24, 19)
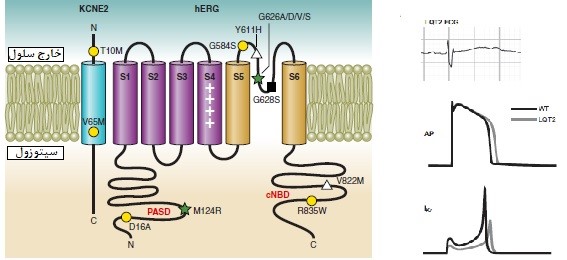
شکل9: نقص کانال یونی در بروز LQT2 و اثر آن بر الکتروکاردیوگرام (19,24)
LQT2: یکی دیگر از انواع شایع آریتمی است که بیماران مبتلا به این نوع LQT حدود 45 درصد از کل بیماران مبتلا به LQTS مادرزادی را تشکیل میدهند. سن متوسط بروز، به دلیل یک رویداد قلبی، 12 سال است. این بیماران داری جهش در ژنی به نام hERG هستند. ژن hERG (با نامKCNH2 هم شناخته میشود) کدکننده پروتئین شناخته شدهای به نام KV11.1 است که زیر واحد α را در کانال پتاسیم تشکیل میدهد (24). زیر واحدهای KCNH2 کمپلکسی را با KCNE2 که یک پروتئین غشایی همراه شده با KCNE1 است، تشکیل می¬دهند تا جریان پتاسیم ایجاد شود و قطبیت غشای سلول برقرار شود. این امر موجب بروز پتانسیل عمل قلب میگردد که به هماهنگی ضربان قلب کمک میکند (20). وقتی توانایی این کانال در انتقال جریان الکتریکی در غشای سلول مهار شود یا با استفاده از داروها یا با جهشهای نادر در برخی از خانوادهها, عملکرد خود را از دست بدهد، میتواند منجر به یک اختلال بالقوه کشنده به نام سندرم LQT شود. بیماران مبتلا به LQT2 با کانالهای ناکارآمدی در مقایسه با بیمارانی که تعداد کانالهای طبیعی کاهش یافته را نشان میدهند، بیشتر در معرض آریتمی قرار دارند (شکل9)(25,26). مطالعات تجربی نشان داده است که سرکوب جریان پتاسیم، لزوماً میانگین پتانسیل عمل را طولانی نمیکند، اگرچه باعث افزایش قطبی شدن غشای سلول میشود، زیرا در این حالت، سلولهای قلبی، مدت زمان پتانسیل عمل طولانیتری را پس از سرکوب کانالهای پتاسیم، نسبت به سایر سلولهای سندرمهای QT طولانی نشان میدهند. در این بیماران، بهطور موقت پتانسیل عمل طولانی در سلولهای قلبی ایجاد میشود، بنابراین پراکندگی سطحی دپلاریزاسیون بیش از حد طبیعی رخ میدهد، اما نه به اندازه افزایشی که در بیماران LQT1 مشاهده میگردد (شکل9) (24).
LQT3: این نوع آریتمی، حدود 5 درصد از کل LQTS را شامل میشود. در واقع ناشی از جهش در ژن کدکننده کانال سدیم قلب میباشد. تا به امروز، 9 جهش مجزا که معمولاً شامل جایگزینیهای اسید آمینه یا حذف در بخشهایی است که در دامینهای III و IV کانال قرار دارند، گزارش شده است. همه این جهشها باعث تغییر قابل توجهی در ویژگیهای پروتئین کانال Na+ میشوند که به طور مستقیم یا غیرمستقیم باعث افزایش مجدد قطبیت بطنی میشود (شکل10) (24). ژن SCN5A، زیر واحد α کانال یون سدیم قلب یا NaV1.5 را کد میکند که یا به عنوان یک مونومر عمل میکند یا به عنوان یک دیمر در کمپلکس کانال یونی مونتاژ میشود. جهشهای افزایش عملکرد SCN5A منجر به اختلال در روند غیرفعال شدن سریع کانالهای سدیم قلب می گردند و با فنوتیپ LQT3 مرتبط هستند و 5 تا 10 درصد از کل موارد LQTS را تشکیل میدهند. LQT3 ممکن است بر روی الکتروکاردیوگرام سطحی به عنوان یک فاصله طولانی ایزوالکتریک قبل از موج T نسبتاً طبیعی ظاهر شود. این زیرگروه LQTS کمترین پاسخ را به مسدود کنندههای بتا میدهد و در عین حال کشندهترین نوع آریتمی است. بیش از 300 نوع جهش در ژن SCN5A مربوط به LQT3 شناخته شده است. از نظر بالینی، حوادث آریتمی LQT3 اغلب با اختلال برادی کاردی قلب همراه است، بنابراین بیماران LQT3 همانطور که در شکل 10 نشان داده شده است، با آریتمیهای بدخیم حتی در حالت استراحت و در طی خواب شبانه ظاهر میشوند. برخلاف بیماران مبتلا به جهش LQT1، بیماران LQT3 در حین ورزش در معرض خطر نسبتاً کمی قرار دارند، زیرا که در ضربان قلب سریع، Na+ در سلول تجمع مییابد و گرادیان Na+ را در سراسر غشا و در نتیجه مقدار جریان Na+ داخلی را کاهش میدهد (شکل10) (22,24,26). در شکل11 محرکهای آریتمی قلبی در LQT1، LQT2 و LQT3 با ورزش، احساسات و استراحت بررسی شده است. همانطور که در شکل مشخص شده است، بروز LQT1 بیشتر در هنگام ورزش بوده است و بروز LQT2 وLQT3 بیشتر در هنگام خواب و استراحت اتفاق میافتند (20).
نقش کانالهای یونی در LQTS: تاکنون مشخص شده که حدود 95% از موارد LQTS در نتیجه جهش در ژنهای کانال پتاسیم ایجاد شدهاند و ژنهای کانال سدیم (LQT3) تنها 5-4% موارد را شامل میشوند (22). نکته مهم این است که برای تقریبا 30-40% از موارد بیماری، تاکنون جهش ژنی شناسایی نشده است و احتمال دارد جهشها در نواحی غیر کدشونده ژنها و یا در ژن های تنظیمی رخ دهند و یا اینکه تنها ژنوم هستهای در بروز بیماری دخالت نداشته باشد (14,19,24,27). کانالهای سدیم نرمال معمولا یک یا دوبار در طول دپولاریزاسیون باز هستند و سپس وارد مرحله غیر فعالشدن سریع میشوند و به ندرت در طول دپولاریزاسیون به حالت باز بر میگردند. در LQT3 حالت غیرفعال شده ناپایدار میشود و سبب بازگشت کانال به حالت باز رخ می دهد. این امر باعث ادامه جریان رو به داخل یونها و طولانی شدن مدت پتانسیل عمل و در نوارهای اکوکاردیوگرام سببLong QT میشود (شکل10). مطالعات اخیر نشان می¬دهد که برخی از بیماران به دلیل داشتن حالت همپوشانی جهشهای ژن SCN5A دارای هر دو فنوتیپ سندرمهای بروگادا و LQT3 هستند (14,19,24). مشخص شده است که تقریباً در 30% از بیمارانLQT ، فاصله QT کاملاً نرمال است. این بیماران را به عنوان ناقلین جهشهای خاموش میشناسند، یعنی افرادی هستند که از نظر ژنتیکی همان نقص را دارند، ولی از لحاظ فنوتیپی هیچ گونه علایم بالینی ندارند و در حدود 15-20% در معرض خطر سنکوپ یا ایست قلبی قبل از سن 40 سالگی هستند. تاریخچه خانوادگی در 60% بیماران، دیده میشود که نشاندهنده نرخ بالای توارث این سندرم است (19,24). یافتههای محققین روی تعداد زیادی از بیماران ثابت میکند که بروز آریتمی قلبی در LQTS در حالتهای مختلف بیماری، متفاوت است. بیماران LQT1 دارای جهش¬های فقدان عملکرد (Loss of function)، روی ژن کانال پتاسیم هستند که در خلال ورزش و حرکات بدنی و شنا بیشتر بروز مییابد. در LQT2 بیماران دارای جهشهای فقدان عملکرد روی ژن KCNH2 هستند که در بیان کانال پتاسیم نقش دارد و اثر آنها تحت شرایط استرس های هیجانی دیده میشود (22). بیماران LQT3 دارای جهشهای کسب عملکرد (Gain of function) در ژن کانال سدیم هستند که غالبا در حالت استراحت باعث آریتمی میشود (شکل11) (19,22,24). این اطلاعات از این رو دارای اهمیت است که میتوان به بیماران حالتهای خطرزا را معرفی کرد. به عنوان مثال از بیماران یا کودکان مبتلا خواست که از شنا و ورزش-های هیجانی پرهیز کنند یا در معرض صداهای بلند حتی صدای زنگ تلفن یا ساعت در زمانهای استراحت قرار نگیرند. طی مطالعات انجام شده و اطلاعات به دست آمده، جزئیات هر کدام از انواع LQTs بحث شده در جدول 2 آورده شده است (19,24).
پاتوفیزیولوژی سندرم LQT: علامت مشخصه LQTs روی ECG، طویل شدن فاصله QT و تغییراتی در شکل موج T است (17). این محدوده QTc در بیماران از 410 تا بالاتر از 600 msec (میلی ثانیه) میباشد. در جمعیت نرمال این محدوده از 350 تا 460msec است. همپوشانی در ناحیه 410 تا 460 بین افراد بیمار و سالم، تشخیص این سندرم را بسیار مشکل می¬کند، مخصوصاً اینکه، 30% ناقلین دارای QTc بین 410 تا 460 msec هستند. ریسک فاکتورهای محیطی در بروز LQT عبارتند از ورزش و حرکات بدنی شدید بهویژه دویدن و شنا، اضطراب و استرس، خشم و عصبانیت و از جا پریدن حتی به دلیل شنیدن صدای زنگ تلفن یا ساعت و ترس، که در بیشتر از 90% موارد مسبب بروز آریتمی قلبی در بیماران میشود (17). تشخیص LQTS از طریق مشاهده طویل شدن فاصله QTc در اکوکاردیوگرام بیمار و وجود سابقه خانوادگی در بروز سندرم میباشد، اما در اغلب موارد، تشخیص سندرم با مشکلاتی مواجه است. از این رو محققی به نام شواترز (Schwartz) و همکارانش در سال 1993، از روش امتیازدهی برای تشخیص بیماران استفاده کردند. این امتیازات براساس مشاهدات اکوکاردیوگرام، سابقه بالینی، تاریخچه خانوادگی و علایم بیمار، طراحی شده است (جدول 3). بر مبنای این سیستم امتیازدهی، احتمال LQTS مطابق رابطه زیر است: امتیاز کمتر از 1با احتمال LQTs کم، امتیاز بین 1/5 تا 3 با احتمال بروز متوسط و امتیاز بالاتر از 5 دارای احتمال بالای بروز LQTs میباشد.
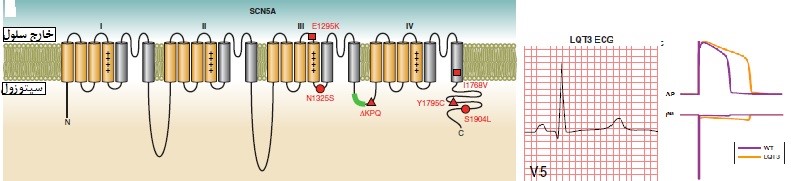
شکل10: نقص کانال یونی در بروز LQT3 و اثر آن بر الکتروکاردیوگرام (24, 19)
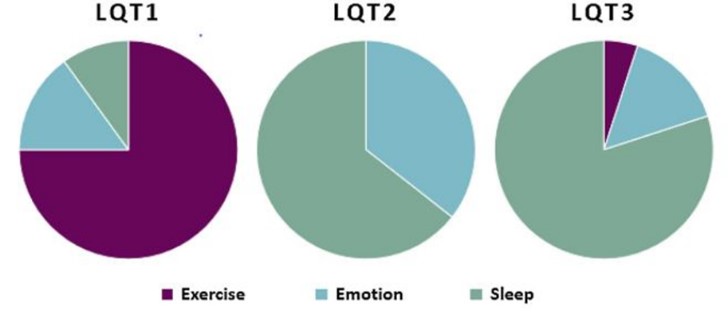
شکل11: محرکهای آریتمیهای قلبی درLQT1 ، LQT2 و LQT3 (20)
جدول 2: تفکیک و تشخیص LQT1, LQT2, LQT3 از یکدیگر بر اساس جهشهای شایع (19)
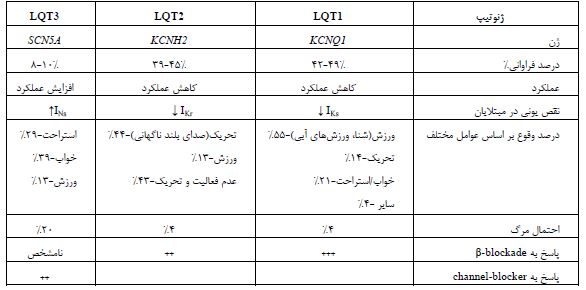
جدول 3: مشاهدات الکتروکاردیوگرام و سیستم امتیاز دهی شوارتز و همکارانش (20)
رویکردهای درمانی بیماری LQTs: طبق مطالعات انجام شده محققان نشان دادهاند که درمان مبتنی بر RNAi میتواند در درمان بیماری LQTs موثر باشد. در جهشهای منفی غالب LQTs از RNAi برای خاموش کردن محصول پروتئینی استفاده میشود. لو و همکاران اثر RNAi را بر جهشهای منفی غالب LQT2 در موقعیت E637K در ژن hERG مورد بررسی قرار دادند. در این روش با استفاده از siRNA قسمت خاصی از توالی را در موقعیت E637K-hERG مورد هدف قرار دادند و عملکرد پروتئین را اصلاح کردند. این تکنیک اثرات جهش E637K-hERG را کاهش داد به طوریکه خواص کینتیکی پروتئین جهش یافته در حد سطوح پروتئین نرمال افزایش یافت (20,28). با این حال، مشخص است که اگر این فناوری در داخل بدن و در صورت کافی بودن یک کپی از پروتئین نرمال کار کند، پس وجود همان یک کپی از ژن برای حفظ عملکرد طبیعی در داخل بدن کافی است. یکی دیگر از روشهای درمانی، توانایی تولید سلول های بنیادی پرتوان القایی (iPSC)است که زمینه تحقیقات قلب و عروق را متحول کرده است، امکان تجزیه و تحلیل عملکردی بافت قلب بیمار در این روش فراهم میشود. فناوری سلولهای بنیادی به محققان این امکان را میدهد که یک نمونه خاص بیمار را از فیبروبلاستهای پوست از یک خانواده تولید کنند که در نهایت منجر به ایجاد جریان الکتریکی درون سلولی و بین سلولی در بیماران میشود (29). تکنیک دیگر، کاربرد CRISPR/Cas9 میباشد که یک تکنیک ویرایش ژنوم بسیار دقیق و کارآمد است که سریعتر و ارزانتر از سایر ویرایشهای ژنی قبلی است. این تکنیک میتواند خطوط جهش یافتههای ایزوژنیک را تولید کند به این صورت که با کنترلiPSCها یا ایجاد iPSCهای اصلاح شده ژنتیکی مربوط به نواحی جهش یافته میتوانند باعث از بین بردن تفاوت های اپی ژنتیکی یا ناشناخته شوند و تنوع فنوتیپی بیماری LQTs را اصلاح کنند. با اینحال تکنیکهای گفته شده دارای محدودیتهایی نیز هستند و چنانچه مورد نظر محققان باشند، لازم است اصلاحاتی در روشها صورت گیرد تا کارآیی آنها افزایش یابد (30). پیشرفتهای اخیر پیشنهاد میکنند که تکنیکهای RNAi, iPSC, CRISPR/Cas9 میتوانند امیدی برای درمان بیماریهای LQTs به شمار آیند (20).
سندرم QT کوتاه (SQT): سندرم SQT بیماری وخیم با الگوی توارثی اتوزوم غالب با نفوذ فنوتیپی بالا میباشد که در هر دو جنس و در تمام سنین با علائمی از جمله کوتاه شدن بازهQT ، فیبریلاسیون دهلیزی، آریتمی و تاکی کاردی بطنی همراه است که منجر به سنکوپ و مرگ ناگهانی قلبی میشود (31). دپولاریزاسیون سریع قلب موجب کاهش فاصلهQT بر روی ECG شده که زمینه ایجاد آریتمیهای قلبی که منجر به سنکوپ و مرگ ناگهانی میشود را بهوجود میآورد (34-32). بسیاری از مبتلایان در طول زندگی خود هرگز علائمی نداشتند و یا حتی اولین تجربه آنها، مرگ ناگهانی قلبی بوده است، به همین علت این بیماری مرگبار در اکثر موارد در آریتمیهای وخیم و همچنین نتایج مرگبار آن در جوانان تشخیص داده شده است (31,32).
اساس ژنتیکی و اهمیت کانالهای یونی: بیشتر از 30 جهش نادر در 8 ژن برای سندرم SQT که میتواند مادرزادی یا اکتسابی باشد، شناسایی شده است که بر حسب زیرگروههای SQTs ژنهای CACNA1C, CACNA2D1, CACNB2, KCNH2, KCNJ2, KCNQ1 مشخص شدهاند که در جدول4 آورده شده است (35,36):
با توجه به توضیحات جدول 4، جهش ها در مجموع، دو عملکرد مهم و حیاتی را شامل میشوند (35,36):
1-افزایش عملکرد پروتئین در ژنهای کدکننده کانالهای بیرون بر پتاسیم
2-کاهش عملکرد در زیرواحدهای متفاوت کانالهای کلسیم type-L قلبی و همچنین 2 ژن SLC4A3 و SCN5A که در جدول 5 بر حسب فنوتیپ با سایر ژنها آورده شده است (35): در جدول 5 دسته بندی جهشها آورده شده است که بهصورتزیر می توان آن را توضیح داد (35):
الف. جهشهای مرتبط با سندرم SQT
1- ژنهای کدکننده کانال پتاسیم KCNQ1, KCNj2, KCNH2
2- ژن SLC4A3 (35)
ب. جهشهای مرتبط با سندرم بروگادا که کوتاه شدن بازه QT را نشان می¬دهد، اما ثابت کنندهی تشخیص قطعی ابتلا به سندرم SQT نیست:
1- ژنهای کدکننده کانال کلسیم CACNB2, CACNA2D1, CACNA1C
2- ژن SCN5A (35,37)
همچنین جهش در 5 ژن دیگرKCNH2, KCNQ1, KCNJ2, CACNA1C, CACB2b نیز در افراد مبتلا یا مشکوک به سندرم SQT به دلیل کشنده بودن بررسی شده است (33). همانطور که در شکل 12 هم نشان داده شده است، ژنهای مختلفی که جهش در هر کدام از آنها منجر به بروز انواع آریتمی میشود، با هم همپوشانی دارند (33). اما چه تغییراتی در جریانهای یونی باعث کوتاه شدن بازه QT میشود؟ درواقع کاهش جریآنهای قطبش مجدد به سمت داخل (repolarizing inward)و یا همچنین افزایش جریانهای قطبش مجدد به سمت خارج (repolarizing outward ) باعث قطبش مجدد زودهنگام میشود. این قطبش مجدد زودهنگام، خود باعث کوتاه شدن دوره پتانسیل عمل و کوتاه شدن بازهQT می¬گردد. همانطور که در شکل 13 نشان داده شده است، جهشهای کسب عملکرد پتاسیم و جهشهای از دست دادن عملکرد کانالهای کلسیم و سدیم، منجر به یک فاز رپلاریزاسیون کوتاه شده در طول پتانسیل عمل و در نهایت منجر به کوتاه شدن فاصله QT میشود (33).
تشخیص SQTS: تشخیص سندرم SQTs بر اساس تاریخچه خانوادگی، نشانههای سنجش بیماری و نمودار الکتروکاردیوگرام بیمار انجام میشود (34,37). تشخیص بالینی بیماری با پیگیری حالتهای مختلف در بیمار انجام میگیرد که عبارتند از سابقه تپش قلب و سنکوپ، تاریخچه خانوادگی سنکوپ و مرگ ناگهانی یا بروز مرگ در سنین کم در خانواده. همچنین علتهای ثانویه بروز بیماری نیز باید مورد بررسی قرار گیرند، مانند: هیپرکلسمی، هیپرکالمی، تب شدید و اسیدوز. شوارتز و همکاران سیستم امتیازدهی خاصی را برای کمک به تشخیص سندرم SQTS پیشنهاد کردهاند که طبق جدول 6 دستهبندی می¬شود (37):
طبق جدول 6، امتیاز کمتر از 2 با احتمال کم بروز بیماری، امتیاز 3 با احتمال متوسط بروز بیماری و امتیاز بیشتر از 4 با احتمال بالای بیماری همراه است. اگر مقدار QTCحدود <300-320 ms باشد، طبق مطالعات جدید، بیمار دارای سندرم SQTS است و چنانچه مقدار QTC در محدوده<360 ms باشد در دسته SQTS4/SQTS5 قرار میگیرد. طبق مطالعه¬ای که ویسکین (Viskin) انجام داده است، زنان دارای QTC در محدوده <340 ms و مردان در محدوده <330 ms مبتلا به SQTS تشخیص داده میشوند. در خانوادههایی که دارای سابقه مرگهای ناگهانی قلبی هستند، این محدوده در زنان <370 ms و در مردان <360 ms میباشد (35,38). سندرم بروگادا: سندرم بروگادا (BrS) نوعی بیماری آریتموژنیک توارثی است که با اختلالات هدایت قلبی مشخص میشود و روی نوارهای اکوکاردیوگرام بهصورت بلندشدن قطعه ST (ST-segment elevation) دیده میشود (شکل14) (39,40). این بیماران شدیدا در معرض مرگ های ناگهانی قلبی (SCD) هستند که در نتیجه فیبریله شدن بطنی (Ventricular fibrillation) رخ میدهد (41). سندرم بروگادا عمدتاً در دوران بزرگسالی بروز میکند و متوسط سن مرگ ناگهانی تقریباً ۴۰ سال است. درواقع سندرم بروگادا، ناشی از اختلال کانال سدیم با اختلالات هدایت پیشرونده وابسته به سن، مانند طولانی شدن PQ ECG، QRS، و بازههای HV مرتبط است. اختلال در جریان سدیم به بلوکه شدن هدایت موضعی در اپیکاردیوم کمک میکند و منجر به چندین قله در کمپلکس QRS و تحریک فیبریلاسیون دهلیزی و بطنی میشود (39,42,43).
تاریخچه و ژنتیک سندرم بروگادا: در سال 1992 پدرو و جوزف بروگادا (Pedro and Josef Brugada)، برای اولین بار سندرم بروگادا را به عنوان یک بیماری توارثی معرفی کردند که باعث مرگ ناگهانی در خانوادهای میشد که دارای تاریخچه فامیلی از بروز چنین مرگهایی بودند (44,45). پس از آن در سال 1998 مشخص شد که جهشهای ژن SCN5A روی کروموزوم 3p21 با سندرم بروگادا ارتباط دارند. اگرچه تنها در 15-30% موارد جهشهای این ژن، مسئول بروز سندرم بروگادا شناخته شده است و در بقیه موارد، با وجود اینکه اغلب آنها خانوادگی هستند، ژن یا جایگاه کروموزومی خاصی تاکنون شناسایی نشده است (43,46). اولین ژن مرتبط به سندرم بروگادا، SCN5A است. جهشها در این ژن مسئول بروز LQT3 نیز هستند. تاکنون حدود 60 جهش در این ژن شناسایی شده است و تقریباً 24 جهش از میان آنها در سیستمهای بیانی مطالعه شدهاند و مشخص شده که اثر آنها بهصورت فقدان عملکرد (Loss of function) میباشد و به 4 حالت ممکن است عمل کنند (46):
1) باعث عدم بیان ژنهای کدکننده کانال سدیم میشوند.
2) کانالهای سدیم وابسته به ولتاژ و زمان هستند، یعنی در یک چرخه فعالشدن، غیرفعال شدن و دوباره فعالشدن، عمل میکنند. جهشها ممکن است این چرخه را دچار شیفت یا تغییر کنند.
3) جهشها باعث میشوند کانال سدیم وارد حالت حد واسط و غیرفعالی شود که خود سبب حرکت آهسته یونها میگردد.
4) جهشها گاه به طور خیلی سریع و برگشت ناپذیر، کانال سدیم را غیرفعال میکنند (40).
همانطور که در شکل 15 نشان داده شده است، رابطه بین جهشهای ژنی و کانالهای یونی و عملکرد آنها با سندرم بروگادا مشخص شده است (41).
جدول 4: زیر گروههای SQTS و ژنهای دخیل در بروز بیماری (35)
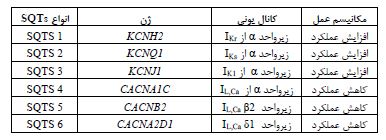
جدول5: ژنهای مرتبط با سندرم SQT و SQT interval
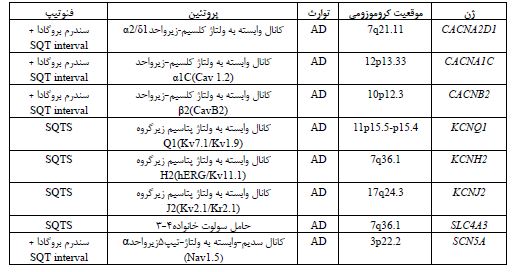
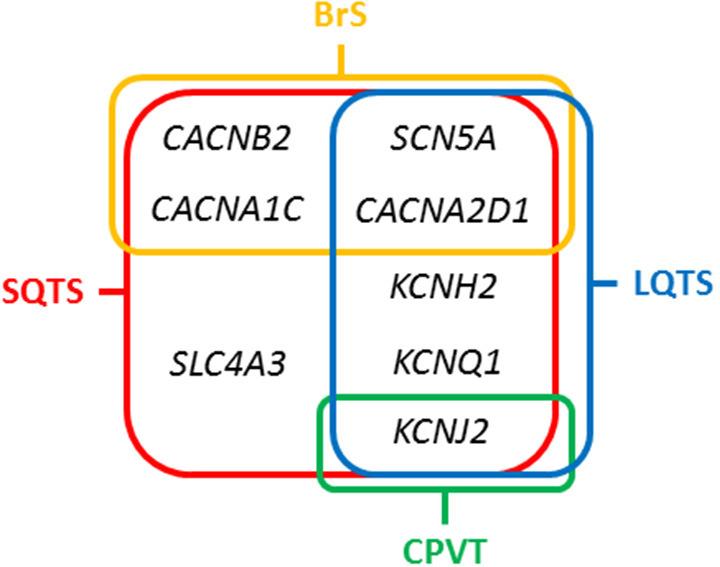
شکل 12: همپوشانی ژنهای مرتبط با LQTS, SQTS و سندرم بروگادا (33)

شکل13: بررسی جهشهای کسب عملکرد و از دست دادن عملکرد در کانالهای یونی و تاثیر در فاصله QT (33)
جدول6: امتیازدهی شوارتز برای تشخیص سندرم SQTS (35)
.JPG)
.jpg)
شکل14: دیاگرام ECG بیماران سندرم بروگادا. در این شکل بلند شدن قطعه ST در بیماران مشخص است (1)
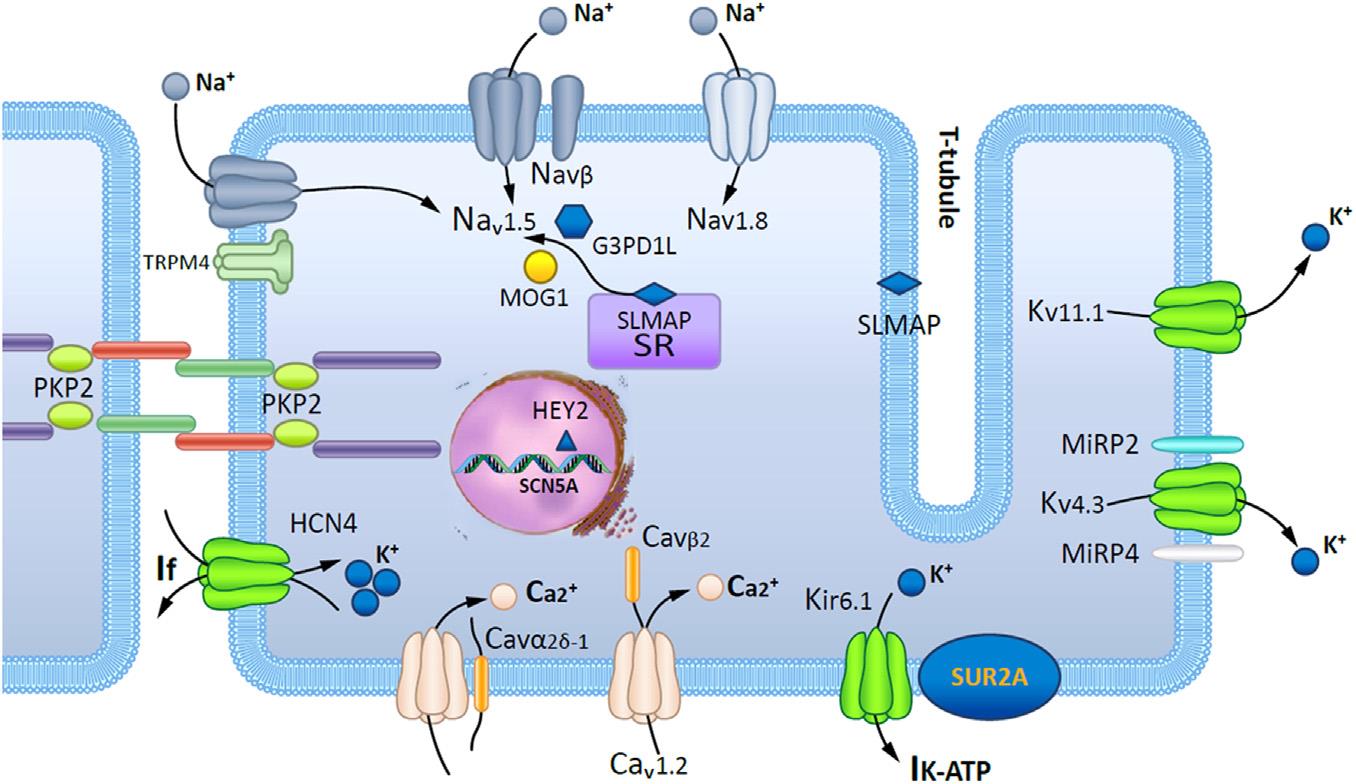
شکل15: نمایش شماتیک یک کاردیومیوسیت که پروتئینهای دخیل در پاتوژنز سندرم بروگادا را نشان میدهد (41).
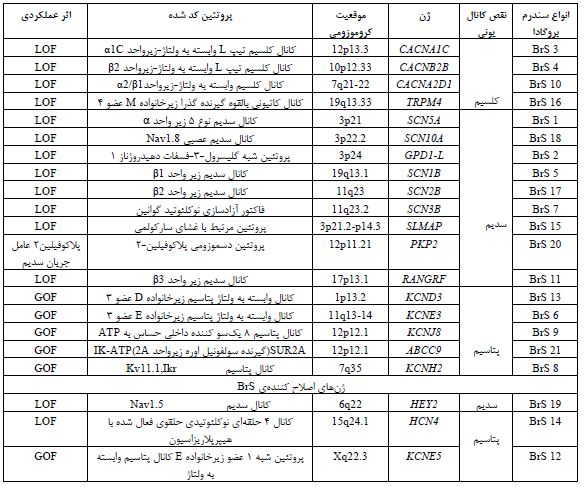
وراثت سندرم بروگادا: در بیشتر موارد، سندرم بروگادا به روش اتوزومال غالب با نفوذ ناقص به ارث میرسد، به استثنای سندرم بروگادایی که وابسته به KCNE5 است که به روش وابسته به X به ارث میرسد. درواقع بیشتر افرادی که مبتلا به سندرم بروگادا هستند، والدین مبتلا دارند که نسبت موارد ایجاد شده توسط یک واریانت بیماریزای جدید، 1% تخمین زده میشود. هر کودک از یک فرد مبتلا به سندرم بروگادای اتوزومال غالب، ۵۰% شانس به ارث بردن واریانت بیماریزا را دارد. تست پیش از تولد برای حاملگیهای پرخطر در صورتی امکانپذیر است که الل بیماری زا در خانواده شناخته شده باشد (41). اگر الل بیماریزا در DNA هیچ یک از والدین شناسایی نشود، ممکن است که یک الل بیماریزای جدید یا موزائیسم سلولهای جنسی در یک والد رخ داده باشد (البته تا به امروز، موزائیسم سلولهای جنسی در سندرم بروگادا توصیف نشده است). اگر چه بیشتر افرادی که مبتلا به سندرم بروگادا تشخیص داده میشوند، الل بیماریزا را از یک والد به ارث بردهاند، تاریخچه خانواده ممکن است به دلیل عدم تشخیص اختلال در اعضای خانواده، نفوذ ناقص، مرگ زودهنگام والدین قبل از شروع علائم، یا شروع دیر هنگام علائم در والدین مبتلا، منفی به نظر برسد (47-42). تشخیص براساس یافتههای بالینی و یا به وسیله شناسایی یک واریانت اللی بیماری¬زای هتروزیگوت (یا هموزیگوت در5 KCNE) در یکی از ۲۳ ژن زیر است:ABCC9, CACNA1C, CACNA2D1, CACNB2, FGF12, GPD1L, HCN4, KCND2, KCND3, KCNE5, KCNE3, KCNH2, KCNJ8, PKP2, RANGRF, SCN1B, SCN2B, SCN3B, SCN5A, SCN10A, SEMA3A, SLMAP, and TRPM4. (42). مطالعات مختلف نشان میدهند که هیچ فنوتیپ دیگری با واریانت بیماریزای ایجاد کننده سندرم بروگادا که در GPDL1،HCN4 و RANGR شناسایی شدهاند، مرتبط نیست. تعدادی از این ژنها در (جدول7) آورده شده است (41):
جدول7: ژن¬های مرتبط با سندرم بروگادا (41)
SCN5A: رابطه بین ژن بیماری¬زای SCN5A و سندرم بروگادا در سال ۱۹۹۸ شناسایی شد، ژن SCN5A به عنوان اولین ژن دخیل در سندرم بروگادا شناسایی شد که متداولترین ژن مرتبط با این سندرم نیز معرفی شده است، با این وجود، بیش از 10 سال است که ژن¬های مرتبط با این سندرم در حال شناسایی هستند (44). تاکنون بیش از ۱۰۰ نوع جهش بیماریزای مختلف در SCN5A گزارش شدهاند که تقریبا نیمی از آنها از نظر بالینی تایید شدهاند (46). چندین نوع جهش بیماریزای مختلف که بر ساختار، عملکرد و عبور از کانالهای سدیم تاثیر میگذارند نیز شناسایی شدهاند (42). توالی ژنومی این ژن، شامل بیش از ۱۰۰ کیلوباز طول دارد. این ژن شامل ۲۸ اگزون است. ژن SCN5A زیر واحد آلفا کانال سدیم قلب را رمزگذاری میکند و مسئول فاز صفر پتانسیل عمل قلب است. پروتئین آن حاوی ۲۰۱۶ اسید آمینه است که شامل چهار تکرار داخلی است، هر کدام از آنها دارای پنج بخش هیدروفوبیک (S۱، S۲، S۳، S۵، S۶) و یک بخش دارای بار مثبت (S۴) میباشند. بخش S۴ احتمالاً سنسور ولتاژ است و با یک سری اسید آمینه با بار مثبت مشخص میشود (48). این پروتئین غشایی یکپارچه، نفوذپذیری یون سدیم وابسته به ولتاژ را در غشاهای تحریکپذیر تعدیل میکند. با فرض اینکه ترکیبات باز یا بسته نسبت به اختلاف ولتاژ در طول غشا واکنش نشاندهند، پروتئین یک کانال انتخابی سدیم را تشکیل میدهد که ممکن است یونهایNa+ را مطابق با گرادیان الکتروشیمیایی عبور دهند. این کانال مسئول افزایش اولیه پتانسیل عمل در ECG است. این پروتئین در ماهیچه قلب دهلیز و بطن انسان بیان میشود، اما در ماهیچه اسکلتی، مغز، میومتر، کبد و یا طحال وجود ندارد (42,48) (شکل16). انواع پاتوژنیک و موتانت در ژن SCN5A منجر به کاهش جریانNa+ توسط یکی از دو مکانیزم اصلی در سلول میشود: عدم بیان کافی از کانال تغییر یافته یا غیرفعال شدن سریع کانال که هر دو حالت منجر به تغییر در پتانسیل عمل سلولهای قلب میگردد که در نهایت منجر به آریتمی میشود (46,42).
نقش کانالهای یونی در بروگادا: چون ژن SCN5A کدکننده زیر واحد α کانال سدیم قلب است و جهش در آن باعث کاهش جریان سدیم دپولاریزه کننده و بروز علایم سندرم بروگادا میشود، این سندرم را به عنوان نوعی بیماری کانال یونی (Channelopathies disease) میشناسند (46). در سال 1995 مشخص شد، ژن SCN5A مسئول بروز گروهی از بیماران LQTS (LQT3) نیز هستند که گروه نسبتاً نادری از LQTSها را شامل میشود. جهشهای این ژن در LQTS، باعث غیرفعال شدن جریان سدیم (یک جریان دپولاریزهکننده) در طی فاز پلاتو (Plateau phase) پتانسیل عمل، یا به عبارت دیگر، کسب عملکرد (Gain of function) زیاد کانال میشود که منجر به طولانی شدن رپلاریزاسیون و بنابراین حالتLong QT میشود (46). دیگر جهشهای این ژن باعث بروز علایم بالینی متفاوتی مانند بالارفتن موج ST در سندرم بروگادا میشود، اما در سندرم بروگادا، جهشهای اخیر سبب فقدان عملکرد کانال میگردد. بنابراین میتوان گفت جهشهای ژن SCN5A منجر به بروز فنوتیپهای کلینیکی هتروژنوس و متفاوتی میشود. اما در همه بیماریهای ایجاد شده توسط این جهشها (LQTS، سندرم بروگادا، بیماری غده سینوسی و بیماریهای انتقال یونی) مرگ ناگهانی، غالباً در خواب و استراحت رخ میدهند که محققان دلیل آن را به هم خوردن تعادل و هماهنگی بین ایمپالسهای سمپاتیک و پاراسمپاتیک میدانند که مهمترین عامل بروز آریتمی است (شکل16) (46).
میزان شیوع سندرم مرگ شبانه: به دلیل کم بودن اطلاعات ژنتیکی موجود در این سندرم، میزان نفوذ بیماری در میان جمعیتها، هنوز به طور دقیق تعیین نشده است. تخمین محققان 10000/ 5-1 نفر در کشورهای غربی است. اما شیوع بالای سندرم (2500/1) در کشورهای شرقی بهویژه تایلند نیز دیده میشود. در جنوب آسیا مانند فیلیپین، ژاپن و تایلند، چون این سندرم باعث مرگ در حالت خواب و در نیمههای شب میشود، از این سندرم به عنوان مرگ شبانه بدون توضیح و ناگهانی (Sudden Unexplained Nocturnal Death) یاد میکنند که بیشتر در افراد جوان رخ میدهد (40). توارث سندرم بروگادا، اتوزومال غالب است و بیشتر بهصورت تاکی کاردیای بطنی چند شکلی (Polymorphic ventricular tachycardia) و مرگ ناگهانی در بیماران جوان و در درجه کمتر در نوزادان و کودکانی دیده میشود که دارای قلبهای سالم از نظر ساختاری هستند. اگرچه میانگین سنی در زمان اولین تشخیص یا مرگ ناگهانی قلبی، حدود 40 سالگی است، اما فنوتیپ بروگادا در محدوده سنی وسیعی گزارش شده است. این سندرم مسئول 20% از مرگهای ناگهانی در بیمارانی با قلبهای سالم است (40). در اکوکاردیوگرام این بیماران علاوه بر بلندشدن قطعه ST، طولانی شدن فاصله QT هم دیده میشود. با این وجود در برخی از بیماران الگوی اکوکاردیوگرام هم در تشخیص بیماری ناتوان است (49). مشخصات کلی این سندرم در جدول 8 ذکر شده است. در LQTs، سن وقوع اولین علایم بیماری، دهههای اولیه زندگی است، ولی در سندرم بروگادا، این علایم در دهه چهارم حیات فرد بیشتر دیده میشود. مشخص شده است که سندرمهای LQTs و بروگادا دارای نقصهای ژنتیکی مشابه هستند، اما علایم کلینیکی وابسته به سن متفاوتی را نشان میدهند. دلیل این گونه بروزهای متفاوت هنوز مشخص نشده است، ولی احتمال دارد به این دلیل باشد که این سندرمها بهصورت مولتی فاکتوریال یا چند عاملی هستند (49). از میان بیماران گزارش شده با سندرم بروگادا، حدود 90% آنها مرد بودند و میانگین سنی
آنها بین 22 تا 65 سال گزارش شده است. محققین با مشاهده بروز زودتر علایم LQTs و بروگادا سندرم، در مردان پیشنهاد میکنند که جنسیت و فعالیت های فیزیکی در مردان در ظهور علایم کلینیکی نقش دارد (49). از فاکتورهای دیگری که در بروز علایم ناگهانی این دو سندرم نقش دارند: تغییرات هورمونی، اختلالات الکترولیتی، داروهای مصرفی بیمار، میزان جذب غذا، تب شدید و سن را می توان نام برد. هیچ کدام از این فاکتورها مستقیما در افزایش وخامت علایم بالینی نقش ندارند و بنابراین بهتر است از واژه تعدیل کننده (Modifier) برای آنها استفاده شود. با این وجود جهشهای شناسایی شده، تنها 30 تا 40% از موارد سندرم را توجیه میکنند و تاکنون بقیه عوامل دخیل در بروز سندرم بروگادا ناشناخته مانده است. از این رو یکی از احتمالات محققین، دخیل بودن ژنوم میتوکندری و وجود اختلالات زنجیره تنفسی، در بروز این سندرم است (48,50).
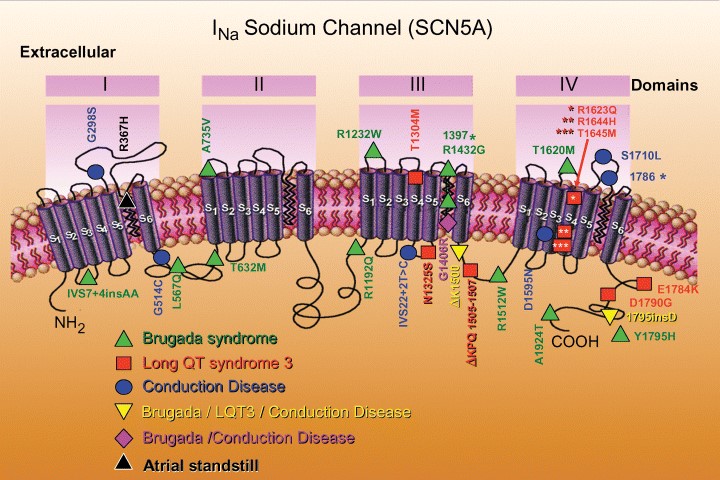
شکل16: بررسی کانال سدیمی SCN5A و عملکرد دومین پروتئینی در بروز سندرم بروگادا (48)
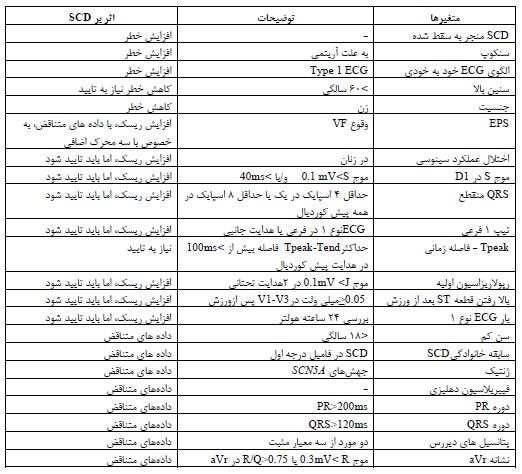
جدول 8: متغیرهای شناسایی شده مرتبط با سندرم بروگادا و مرگ ناگهانی قلبی(SCD) (49)
نتیجهگیری
درک فعلی ما از پاتوفیزیولوژی اختلالات آریتموژنیک قلبی در 20 سال گذشته، عمدتاً از تحقیقات گسترده در مورد کانالوپاتیهای یونی، به ویژه LQTS مادرزادی و سایر موارد مانند سندرم بروگادا به دست آمده است. درک بهتر محققین از عملکرد هر کانال یونی در رابطه با جهشهای ژنی، و نحوه اثر هر تغییر ژنتیکی در عملکرد کانالهای یونی قلب، راه را برای تحقیق و توسعه روشهای درمانی هدفمند دارویی و غیردارویی مؤثرتر، مانند ژن درمانی در بیماران هموار کرده است.
حامی مالی: ندارد.
تعارض در منافع: وجود ندارد.
قلب یک عضو عضلانی است که بهطور مداوم، یعنی در حدود 3 بیلیون سیکل در طول حیات یک فرد، خون را به تمام بدن پمپ میکند و دارای دو حفره دهلیزی و دو حفره بطنی میباشد. یک ضربان قلب ساده از انبساط، یعنی زمانی که دهلیزها و بطنها از خون پر شده و انقباض، زمانیکه خون به کل بدن پمپ میشود، تشکیل شده است (1,2). آریتمی به معنی ریتم غیرطبیعی ضربان قلب است، انواع مختلفی از آریتمی وجود دارد که موجب ایجاد ضربان خیلی سریع (تاکیکاردی) یا خیلی آهسته (برادیکاردی) میشود و درنتیجه قلب پمپاژ غیر مؤثری را انجام میدهد. در واقع در اثر اختلال سیستم هدایت الکتریکی طبیعی قلب، بیماریهای آریتمی قلبی ایجاد میشوند (شکل1). آریتمیها شایع هستند و میلیونها نفر در جهان را درگیر کردهاند و به عنوان یکی از علل اصلی مرگهای ناگهانی در آمریکا شناخته شدهاند که سالیانه موجب مرگ ۴۰۰۰۰۰ نفر میشوند. فیبریلاسیون دهلیزی شایعترین شکل آریتمی در افراد مسن در آمریکاست که تقریباً 2/5 میلیون نفر به آن مبتلا هستند (3،4).
امواج پلاریزه و دپلاریزه: انقباضات منظم قلب وابسته به یک شبکه الکتریکی است که امواج الکتریکی را به تمام قلب هدایت میکند. ضربانساز غالب قلب، گره سینوسی دهلیزی، آغازگر موج دپولاریزاسیونی است که به شکل یک موج گسترش یافته و دهلیزها را برای انقباض تحریک میکند. این گره سینوسی در داخل دیواره خلفی فوقانی دهلیز راست قرار دارد و بهطور طبیعی، پالس الکتریکی ایجاد شده از طریق رشتههای عضلانی به تمام قلب توزیع میشود. داخل سلولهای قلب، در حال استراحت، بار منفی وجود دارد (سلولها پولاریزهاند)، اما زمانی که با تحریک الکتریکی دپولاریزه میشوند، منقبض میشوند. موج دپولاریزاسیون (مثبت شدن بار داخل سلولها) و مرحله رپولاریزاسیون (برگشت بار منفی داخل سلولها) متعاقب آن، روی اکوکاردیوگرام (ECG) ثبت میگردد (شکل2) (5). تحریک الکتریکی دپولاریزاسیون در داخل دهلیزها منتشر میشود و باعث ایجاد موج P بر روی الکتروکاردیوگرام میشود که نشاندهنده انقباض همزمان دهلیزهاست. تحریک الکتریکی از طریق تارهای انتهایی فیبرهای پورکنژ، دپولاریزاسیون را به سلولهای میوکارد بطنی میرساند. دپولاریزاسیون میوکارد بطنی باعث ایجاد کمپلکس QRS بر روی ECG و انقباض بطنها میشود. پس از هر کمپلکس QRS، یک خط ایزوالکتریک افقی دیده میشود که قطعه ST نامیده میشود و پس از آن یک موج T عریض ظاهر میشود. موج T، نشاندهنده فاز سریع انتهایی رپولاریزاسیون بطنی است که در طی آن رپولاریزاسیون با سرعت و بهطور موثر روی میدهد. از آنجا که سیستول یا انقباض بطنی از آغاز QRS تا پایان موج T به طول میانجامد، فاصله QT (QT interval) از لحاظ بالینی اهمیت زیادی دارد (شکل 3). فاصله QT (QT interval) در اکوکاردیوگرام، نشاندهنده دوره فعالیت و بازگشت میوکاردیوم بطنی است. در واقع فاصله QT، زمانی است که سپری میشود تا بطنها دپولاریزه (شروع QRS) و رپولاریزه (انتهای موج T) شوند و این مدت بسته به میزان تپش قلب دارد (6،7).
عامل ایجاد ایمپالسهای الکتریکی قلب: ایمپالس الکتریکی توسط گرادیانت الکتروشیمیایی موجود در غشاء سلولهای عضله قلبی یا کاردیومیوسیتها ایجاد میشود و به تعادل جریان انتقال یونها در داخل و خارج غشاء سلول وابسته است (8). در حفظ این تعادل، کانالهای یونی از اهمیت خاصی برخوردارند و میتوان گفت، اساس مولکولی الکتروفیزیولوژی قلب، کانالهای یونی هستند (3). در ژنوم انسان، حدود 429 کانال یونی وجود دارد و تقریبا 30 ژن کدکننده کانالهای یونی قلب تاکنون شناسایی شدهاند (9,10).
کانالهای یونی: کانالهای یونی قلب، حاوی پروتئینها و گلیکوپروتئینهایی هستند که در سارکولمای کاردیومیوسیتها واقعاند و تشکیل منافذی در غشاء سلول را میدهند که به یونهای خاصی اجازه میدهند، بر اساس شیب الکتروشیمیایی از غشاء عبور کنند که به این ترتیب باعث تنظیم عملکرد سلول میشوند (8,10). چهار نوع کلی از این کانالها وجود دارد: کانالهای بدون دروازه یعنی همواره باز (Non-gatedchannels): مانند پمپهای سدیم و پتاسیم، کانالهای دارای دروازه (Directly gated channels): مانند کانالهای وابسته به ولتاژ (Voltage-gated channels) و وابسته به لیگاند (Ligand-gated channels)، کانالهای وابسته به پیامبرهای ثانویه (Second messenger gated channels): مانند گیرنده¬های پروتئین G (G-protein receptors) و کانالهای وابسته به ذخیره (Store-operated channels): مانند کانالهای پتانسیل موقتی رسپتور (Transientreceptor potential channels) (11). کانالهای یونی برای طیف وسیعی از عملکردهای فیزیولوژیکی مانند سیگنالهای نورونی، انقباضات عضلانی، هدایت عمل قلب، ترشح هورمونی، تنظیم حجم سلول و تکثیر سلولی ضروری هستند و به همین دلیل کانالهای یونی در بیماریهای زیادی دخالت دارند که اغلب آنها بیماریهای توارثی هستند و در نتیجه جهشهایی در ژنهای کدکننده پروتئینهای کانال ایجاد میشوند (11,12). کانالهای وابسته به ولتاژ عصب و عضله از نظر گسترش ایمپالسهای عصبی و انقباضات عضلانی حائز اهمیت هستند. بهطور مثال، کانالهای سدیم با دپولاریزاسیون غشا سلول، فعال میشوند. در حالت باز، آنها بهطور انتخابی اجازه ورود یونهای سدیم را میدهند. جریان یونها به داخل سلول، تولید دپولاریزاسیون موضعی قوی به نام پتانسیل عمل میکند که باعث باز شدن کانالهای سدیم وابسته به ولتاژ جدیدی میگردد که دپولاریزاسیون را شدت میدهد. پس میتوان گفت کانالهای یونی وابسته به ولتاژ، زمینه ساز پتانسیل عمل در سلولهای عضله قلبی هستند. نقص در هر کدام از این کانالهای یونی منجر به اختلال در پتانسیل عمل سلولهای عضله قلبی و اختلالات اکوکاردیوگرام و باعث ایجاد زمینهای برای بروز آریتمیهای قلبی میشود (13).
بیماریهای قلب: بهطور کلی دو نوع بیماری مهم در قلب وجود دارد:
1) کاردیومیوپاتی که در اثر تغییراتی در پروتئینهای سارکومریک و اسکلتسلولی رخ میدهد.
2) بیماری¬های آریتموژنیک که توسط جهشهایی در کانالهای یونی و پروتئینهای کنترل کننده کانال ایجاد میشوند که به این گونه بیماریها،Cardiac channelopathies میگویند. نظیر: سندرم¬های Long QT (LQTs) و بروگادا (Brugada syndrome) و Short QT (SQTs)، تاکی کاردیای دهلیزی (CatecholaminergicPolymorphic Ventricular Tachycardias) و فیبریلاسیون ایدیوپاتیک (13،14).
آریتمیهای قلبی: بیماریهای آریتموژنیک قلب، عوامل مهم مرگهای ناگهانی قلبی در افراد جوان با قلبهای سالم هستند (6). در سالهای اخیر مطالعات گستردهای روی ژنهای کانالهای یونی قلب انجام شده است، اما هنوز 30 تا 45% موارد آریتمی را نمیتوان با این جهشهای شناخته شده توضیح داد (13,15). آریتمی یا ضربان غیر طبیعی قلب، ممکن است بهصورتتغییر در سرعت و یا نظم ضربانهای قلب باشد. در جریان آریتمی، ضربان ممکن است بیش از حد آهسته، بسیار تند و یا نامنظم باشد. آریتمی ممکن است به شکلهای مختلف تظاهر کند. میتواند بهصورت احساس لرزش یا ریزش در سینه (طپش قلب) همراه با درد سینه باشد. گاه باعث سبکی در سر میشود و یا بهصورت حملاتی همراه با بیهوش شدن است. در مواردی هم آریتمی، علامت مهمی ندارد و بیمار به آن توجهی نمیکند، اما وقتی ضربان قلب به حدی کند یا تند باشد که در عملکرد قلب به عنوان یک پمپ، اختلال ایجاد کند، میتواند بیمار را با خطری جدی مواجه سازد (13,15). تعیین ژنهایی که باعث بروز سندرمهای آریتموژنیک توارثی میشوند، مبنای مطالعات مولکولی است که روش تشخیصی جدیدی را علاوه بر اکوکاردیوگرام در اختیار محققین قرار میدهد (شکل5).
شکل1: مقایسه امواج الکتریکی قلب نرمال و قلب دارای آریتمی (https://www.ashwinihospital.co/ECG.html)
شکل2: دو موج دپلاریزاسیون ( انقباض) و رپلاریزاسیون( استراحت) را در ECG نشان میدهد (5)
شکل3: اندازه گیری فاصلهQT در اکوکاردیوگرام (ECG). دیاگرام نشاندهنده ECG نرمال با موج P ( فعالیت دهلیزی)، مجموعه QRS (فعالیت بطنی و شروع انقباض بطنی) و موج T (رپلاریزاسیون بطنی) است. فاصله QT به طول بین شروع موج Q تا انتهای موج T گفته میشود
شکل4: ساختار کاردیومیوسیت بطنی (11)
شکل 5: تاثیر جهش ژنهای مختلف دخیل در بیماریهای قلبی در منحنی اکوکاردیوگرام (4)
نقصهای متابولیکی در بیماران آریتمی: مشخص شده است که متابولیسم میوکاردیال (Myocardial metabolism) میتواند خود را با تعداد زیادی از سوبستراها وفق دهد، اما الگوی دقیق مصرف سوبسترا بر اساس میزان دسترس بودن، تحویل اکسیژن، حجم و تنظیم فیزیولوژی آنها است. بر این اساس اغلب نقصهای متابولیکی تک ژنی می-توانند باعث اختلال در عملکرد کاردیومیوسیتها و در نهایت آریتمی و مرگ ناگهانی فرد شوند (14,15). چون فعالیت بسیاری از کانالهای یونی وابسته به متابولیسم سلولی است، بنابراین نقصهای ژنوم میتوکندری، ممکن است اثرات متفاوتی در بروز بیماری داشته باشند، مانند تفاوتهای هتروپلاسمی در بافتهای مختلف. از میان این بافتها، قلب بیشترین آسیبپذیری را نسبت به جهشهای میتوکندری دارد که غالباً بهصورت اختلالات قلبی بروز پیدا میکند (16). این امر به دلیل ارتباط بسیار نزدیک نقصهای متابولیسم و آریتمیهای قلبی است که ممکن است در سنین بلوغ بیشتر دیده شوند (14,15). دلایل متعددی وجود دارد که جهشهای ژنوم میتوکندری ممکن است در سندرمهای مرگ ناگهانی (Sudden death) دخالت داشته باشند که از آن جمله می-توان به فرکانس، شیوع نسبتا بالا و ارتباط جنسیت با وقوع بیماری اشاره کرد که نشاندهنده عدم توارث مندلی در این بیماران است (17). احتمال بالایی وجود دارد که الگوی توارثی بهصورت پلیژنیک بوده و هر دو ژنوم هستهای و میتوکندریایی در بروز آن دخیل باشند (14). اگر چه بروز بیماری در مردان بیشتر از زنان دیده شده است، اما این امر نمیتواند به دلیل توارث وابسته به X باشد، چرا که در خانوادههای درگیر، مادران بیشتر از پدران، علایم بیماری را نشان می¬دهند و این فرضیه با قدرت مطرح میشود که ژنوم میتوکندری و جهشهای آن ممکن است در روند آریتمی دخالت داشته باشند (14).
انواع آریتمی:
1. سندرم Long QT (LQTS)
سندرم Long QT نوعی بیماری رپولاریزاسیون میوکاردیال است. این آریتمی بدخیم ناشی از کانالوپاتی یونی قلب است که منجر به تاخیر در دو قطبی شدن پتانسیل عمل قلب میشود و با طویل شدن فاصله QT روی اکوکاردیوگرام، قابل تشخیص است (19,18). این سندرم در واقع نوعی آریتموژنیک بطنی (Ventricular arrhythmogenic) است که میتواند بهصورت توارثی یا اکتسابی باشد. بیماران LQTs مستعد برای بیماری Torsade de Pointes بطنی (TdP) هستند و بهطور جدی در معرض مرگهای ناگهانی قلبی (SCD)حتی در صورت داشتن قلب به ظاهر سالم میباشند (17). علائم این سندرم در بیماران، شامل حمله قلبی، طپش قلب در طی ورزش یا هیجانات زیاد و سنکوپ میباشد و در شرایطی هم اولین نشانه، ایست قلبی (Cardiac arrest) است. در موارد اکتسابی، مهمترین دلیل بروز LQTs استفاده از برخی داروهای قلبی است. سه اختلال سندرم انکرین-B (Ankyrin-Bsyndrome)، اندرسون تاویل (Andersen-Tawil syndrome) و تیموتی (Timothy syndrome) هم باعث LQT میشوند (17,20). استرس و اضطراب هم در بروز LQTs اکتسابی بیتاثیر نیست و تحقیقات نشان داده که افراد مبتلا به سندرم، موقعیتهای استرسزای بیشتری را تجربه کرده بودند (21). در LQTs اکتسابی هم، اختلال در مکانیسمهای یونی، مشابه با نوع توارثی آن دیده میشود (17). تاریخچه و انواع LQTS: گزارشهایی در مورد بروز سندرم LQT وجود دارد که فرزندان در خانوادههایی دچار مرگ ناگهانی قلبی شده بودندکه بعضاً یا طی ورزش دچار سنکوپ شده بودند یا استرس و فعالیتهای هیجانی را در سن های 4، 5، 8 و 9 سالگی تجربه کرده بودند (17). طویل شدن فاصله QT در ECG آنها کاملاً مشخص بود و توارث بیماری در آنها اتوزومال مغلوب شناخته شد. سندرم مشابه دیگری با علایم مرگ ناگهانی طی ورزش یا استرسهای هیجانی اما با توارث اتوزومال غالب، در مطالعات بعدی گزارش شد. این دو فرم از LQTs توارثی، اکنون به عنوان سندرمهای ژرویل، لانگ- نیلسن (Jervell، Lange-Nielsen syndrome) و رومانو-وارد (Romano-Ward syndrome) شناخته میشوند (19). دو سندرم دیگر هم طی سالهای اخیر شناسایی شدهاند به نام های آندرسن تاویل و سندرم تیموتی که گاه آنها را به عنوان LQT7 نام میبرند (18). میزان نفوذ (Prevalence) LQTs توارثی در آمریکا، حدود 1 فرد از 7000 فرد تخمین زده شده است و شاید هر ساله باعث مرگ ناگهانی 3000 -2000 نفر کودک و جوان شود. نوع رومانو-وارد، حدود 99% موارد (با میزان نفوذ 1:5000 تا 1:10000) را شامل میشود و فرم ژرویل، لانگنیلسن نادرتر است و در کمتر از 1% بیماران گزارش شدهاست (با میزان نفوذ 1:55000 تا 1:200000) (17).
ژنتیک LQTS: مطالعات مولکولی ثابت کرده است کانالهای یونی که فعالیت الکتریکی قلب را کنترل میکنند، در بروزLQTs های توارثی نقش دارند و در این ارتباط، جهشهایی در ژنهای کدکننده کانالهای یونی قلب شناسایی شده است (22). این ژنها را بر اساس ترتیب کشف، نامگذاری می-کنند مانند: LQT1 ،LQT2 و ... این سندرم از نظر ژنتیکی، بیماری هتروژنوسی است که با جهشهای شناخته شدهای در ژنهای کدکننده کانالهای یونی قلب، همراه است که تعدادی از آنها کانال پتاسیم را کد میکنند و یکی کدکننده کانال سدیم (SCN5A) است. محققان تاکنون بیش از 200 جهش را در بیماران LQT یافتهاند. اما در اغلب خانوادهها، جهشهای جدیدی را هم میتوان یافت (23). جدول 1، ژنها، پروتئینهای کد شده و کانالهای یونی مربوطه را نشان میدهد (20-18،14).
اساس ژنتیکی :QTs17 زیرگونه مختلف LQTs وجود دارد که با جهشهای تک ژنی 15 ژن غالب اتوزومال مرتبط هستند (18). در حال حاضر سه ژن اصلی KCNQ1 وKCNH2 و SCN5A وجود دارد که تقریباً 75 درصد این اختلال را دربرمیگیرند و سایر ژنهای کشف شده، در مجموع کمتر از 5 درصد موارد LQTs را تشکیل میدهند (شکل6) (25-24،20). LQTs بر اساس جهشهای مرتبط با 15 ژن غالب اتوزومال، LQT 1-15، به 17 زیرگروه طبقهبندی میشوند. از این رو، جهش در ژنهای KCNQ1، HERG ، SCN5A ، KCNE1، KCNE2 و KCNJ2 به ترتیب باعث شکل LQT1، LQT2، LQT3، LQT5، LQT6 و LQT7 از LQTS میشود. که سه تا از مهمترین آنها را در ادامه بررسی میکنیم (شکل7) (18,20). LQT1: یکی از زیرگروههای شایع این بیماری است. بیماران مبتلا به این نوع LQT1، که حدود 42 درصد از کل بیماران مبتلا به LQTSهای مادرزادی را تشکیل میدهند، معمولاً قبل از 10 سالگی مراجعه میکنند (26). دلیل اصلی ایجاد این بیماری، اختلال در عملکرد ژن KCNQ1 است که کدکننده زیر واحد α کانال پتاسیم دارای ولتاژ (KV7.1) موجود در غشای سلولی کاردیومیوسیتها است. در واقع KV7.1 موتانت، جریان پتاسیم تأخیری را به آرامی فعال میکند (18,20) و هنگامی که با زیر واحدهای نرمال دیگر ترکیب میشود، پروتئینهای کانالی ناکارآمدی را تشکیل میدهند که به طور غیرطبیعی چین خوردهاند و معمولاً تحت تجزیه سریع قرار میگیرند. کانال KV7.1 از چهار زیر واحد α تشکیل شده است که برای برقراری جریان پتاسیم، با زیرواحدهای β ژن KCNE ترکیب میشوند. زیر واحد α ژن KCNQ1 دارای یک دامین سنجش ولتاژ (S1-4)، یک دامین تشکیل منفذ (S5-6) و همچنین دامینهای انتهای N و C درون سلولی است. LQT1 بر روی الکتروکاردیوگرام سطحی به عنوان یک موج T گسترده و متقارن با یک وقفه QTc طولانی مدت ظاهر میشود (21). لازم به ذکر است که ورزش جسمانی و تحریک سمپاتیک باعث تحریک سنکوپ و مرگ ناگهانی در بیماران مبتلا به LQT1 میشود (شکل8)(24).
جدول 1: ژنهای هستهای ایجاد کننده LQTs. فلش رو به بالا (↑) یا پایین (↓) به ترتیب افزایش یا از دست دادن عملکرد پروتئین را نشان میدهد (24،20-18).
شکل6: خلاصه ژنهای مستعد سندرم QT طولانی (25)
شکل 7: ژنهای دخیل در اختلالات آریتمی ژنتیکی (18)
شکل8: نقص کانال یونی در بروز LQT1 و اثر آن بر الکتروکاردیوگرام (24, 19)
شکل9: نقص کانال یونی در بروز LQT2 و اثر آن بر الکتروکاردیوگرام (19,24)
LQT2: یکی دیگر از انواع شایع آریتمی است که بیماران مبتلا به این نوع LQT حدود 45 درصد از کل بیماران مبتلا به LQTS مادرزادی را تشکیل میدهند. سن متوسط بروز، به دلیل یک رویداد قلبی، 12 سال است. این بیماران داری جهش در ژنی به نام hERG هستند. ژن hERG (با نامKCNH2 هم شناخته میشود) کدکننده پروتئین شناخته شدهای به نام KV11.1 است که زیر واحد α را در کانال پتاسیم تشکیل میدهد (24). زیر واحدهای KCNH2 کمپلکسی را با KCNE2 که یک پروتئین غشایی همراه شده با KCNE1 است، تشکیل می¬دهند تا جریان پتاسیم ایجاد شود و قطبیت غشای سلول برقرار شود. این امر موجب بروز پتانسیل عمل قلب میگردد که به هماهنگی ضربان قلب کمک میکند (20). وقتی توانایی این کانال در انتقال جریان الکتریکی در غشای سلول مهار شود یا با استفاده از داروها یا با جهشهای نادر در برخی از خانوادهها, عملکرد خود را از دست بدهد، میتواند منجر به یک اختلال بالقوه کشنده به نام سندرم LQT شود. بیماران مبتلا به LQT2 با کانالهای ناکارآمدی در مقایسه با بیمارانی که تعداد کانالهای طبیعی کاهش یافته را نشان میدهند، بیشتر در معرض آریتمی قرار دارند (شکل9)(25,26). مطالعات تجربی نشان داده است که سرکوب جریان پتاسیم، لزوماً میانگین پتانسیل عمل را طولانی نمیکند، اگرچه باعث افزایش قطبی شدن غشای سلول میشود، زیرا در این حالت، سلولهای قلبی، مدت زمان پتانسیل عمل طولانیتری را پس از سرکوب کانالهای پتاسیم، نسبت به سایر سلولهای سندرمهای QT طولانی نشان میدهند. در این بیماران، بهطور موقت پتانسیل عمل طولانی در سلولهای قلبی ایجاد میشود، بنابراین پراکندگی سطحی دپلاریزاسیون بیش از حد طبیعی رخ میدهد، اما نه به اندازه افزایشی که در بیماران LQT1 مشاهده میگردد (شکل9) (24).
LQT3: این نوع آریتمی، حدود 5 درصد از کل LQTS را شامل میشود. در واقع ناشی از جهش در ژن کدکننده کانال سدیم قلب میباشد. تا به امروز، 9 جهش مجزا که معمولاً شامل جایگزینیهای اسید آمینه یا حذف در بخشهایی است که در دامینهای III و IV کانال قرار دارند، گزارش شده است. همه این جهشها باعث تغییر قابل توجهی در ویژگیهای پروتئین کانال Na+ میشوند که به طور مستقیم یا غیرمستقیم باعث افزایش مجدد قطبیت بطنی میشود (شکل10) (24). ژن SCN5A، زیر واحد α کانال یون سدیم قلب یا NaV1.5 را کد میکند که یا به عنوان یک مونومر عمل میکند یا به عنوان یک دیمر در کمپلکس کانال یونی مونتاژ میشود. جهشهای افزایش عملکرد SCN5A منجر به اختلال در روند غیرفعال شدن سریع کانالهای سدیم قلب می گردند و با فنوتیپ LQT3 مرتبط هستند و 5 تا 10 درصد از کل موارد LQTS را تشکیل میدهند. LQT3 ممکن است بر روی الکتروکاردیوگرام سطحی به عنوان یک فاصله طولانی ایزوالکتریک قبل از موج T نسبتاً طبیعی ظاهر شود. این زیرگروه LQTS کمترین پاسخ را به مسدود کنندههای بتا میدهد و در عین حال کشندهترین نوع آریتمی است. بیش از 300 نوع جهش در ژن SCN5A مربوط به LQT3 شناخته شده است. از نظر بالینی، حوادث آریتمی LQT3 اغلب با اختلال برادی کاردی قلب همراه است، بنابراین بیماران LQT3 همانطور که در شکل 10 نشان داده شده است، با آریتمیهای بدخیم حتی در حالت استراحت و در طی خواب شبانه ظاهر میشوند. برخلاف بیماران مبتلا به جهش LQT1، بیماران LQT3 در حین ورزش در معرض خطر نسبتاً کمی قرار دارند، زیرا که در ضربان قلب سریع، Na+ در سلول تجمع مییابد و گرادیان Na+ را در سراسر غشا و در نتیجه مقدار جریان Na+ داخلی را کاهش میدهد (شکل10) (22,24,26). در شکل11 محرکهای آریتمی قلبی در LQT1، LQT2 و LQT3 با ورزش، احساسات و استراحت بررسی شده است. همانطور که در شکل مشخص شده است، بروز LQT1 بیشتر در هنگام ورزش بوده است و بروز LQT2 وLQT3 بیشتر در هنگام خواب و استراحت اتفاق میافتند (20).
نقش کانالهای یونی در LQTS: تاکنون مشخص شده که حدود 95% از موارد LQTS در نتیجه جهش در ژنهای کانال پتاسیم ایجاد شدهاند و ژنهای کانال سدیم (LQT3) تنها 5-4% موارد را شامل میشوند (22). نکته مهم این است که برای تقریبا 30-40% از موارد بیماری، تاکنون جهش ژنی شناسایی نشده است و احتمال دارد جهشها در نواحی غیر کدشونده ژنها و یا در ژن های تنظیمی رخ دهند و یا اینکه تنها ژنوم هستهای در بروز بیماری دخالت نداشته باشد (14,19,24,27). کانالهای سدیم نرمال معمولا یک یا دوبار در طول دپولاریزاسیون باز هستند و سپس وارد مرحله غیر فعالشدن سریع میشوند و به ندرت در طول دپولاریزاسیون به حالت باز بر میگردند. در LQT3 حالت غیرفعال شده ناپایدار میشود و سبب بازگشت کانال به حالت باز رخ می دهد. این امر باعث ادامه جریان رو به داخل یونها و طولانی شدن مدت پتانسیل عمل و در نوارهای اکوکاردیوگرام سببLong QT میشود (شکل10). مطالعات اخیر نشان می¬دهد که برخی از بیماران به دلیل داشتن حالت همپوشانی جهشهای ژن SCN5A دارای هر دو فنوتیپ سندرمهای بروگادا و LQT3 هستند (14,19,24). مشخص شده است که تقریباً در 30% از بیمارانLQT ، فاصله QT کاملاً نرمال است. این بیماران را به عنوان ناقلین جهشهای خاموش میشناسند، یعنی افرادی هستند که از نظر ژنتیکی همان نقص را دارند، ولی از لحاظ فنوتیپی هیچ گونه علایم بالینی ندارند و در حدود 15-20% در معرض خطر سنکوپ یا ایست قلبی قبل از سن 40 سالگی هستند. تاریخچه خانوادگی در 60% بیماران، دیده میشود که نشاندهنده نرخ بالای توارث این سندرم است (19,24). یافتههای محققین روی تعداد زیادی از بیماران ثابت میکند که بروز آریتمی قلبی در LQTS در حالتهای مختلف بیماری، متفاوت است. بیماران LQT1 دارای جهش¬های فقدان عملکرد (Loss of function)، روی ژن کانال پتاسیم هستند که در خلال ورزش و حرکات بدنی و شنا بیشتر بروز مییابد. در LQT2 بیماران دارای جهشهای فقدان عملکرد روی ژن KCNH2 هستند که در بیان کانال پتاسیم نقش دارد و اثر آنها تحت شرایط استرس های هیجانی دیده میشود (22). بیماران LQT3 دارای جهشهای کسب عملکرد (Gain of function) در ژن کانال سدیم هستند که غالبا در حالت استراحت باعث آریتمی میشود (شکل11) (19,22,24). این اطلاعات از این رو دارای اهمیت است که میتوان به بیماران حالتهای خطرزا را معرفی کرد. به عنوان مثال از بیماران یا کودکان مبتلا خواست که از شنا و ورزش-های هیجانی پرهیز کنند یا در معرض صداهای بلند حتی صدای زنگ تلفن یا ساعت در زمانهای استراحت قرار نگیرند. طی مطالعات انجام شده و اطلاعات به دست آمده، جزئیات هر کدام از انواع LQTs بحث شده در جدول 2 آورده شده است (19,24).
پاتوفیزیولوژی سندرم LQT: علامت مشخصه LQTs روی ECG، طویل شدن فاصله QT و تغییراتی در شکل موج T است (17). این محدوده QTc در بیماران از 410 تا بالاتر از 600 msec (میلی ثانیه) میباشد. در جمعیت نرمال این محدوده از 350 تا 460msec است. همپوشانی در ناحیه 410 تا 460 بین افراد بیمار و سالم، تشخیص این سندرم را بسیار مشکل می¬کند، مخصوصاً اینکه، 30% ناقلین دارای QTc بین 410 تا 460 msec هستند. ریسک فاکتورهای محیطی در بروز LQT عبارتند از ورزش و حرکات بدنی شدید بهویژه دویدن و شنا، اضطراب و استرس، خشم و عصبانیت و از جا پریدن حتی به دلیل شنیدن صدای زنگ تلفن یا ساعت و ترس، که در بیشتر از 90% موارد مسبب بروز آریتمی قلبی در بیماران میشود (17). تشخیص LQTS از طریق مشاهده طویل شدن فاصله QTc در اکوکاردیوگرام بیمار و وجود سابقه خانوادگی در بروز سندرم میباشد، اما در اغلب موارد، تشخیص سندرم با مشکلاتی مواجه است. از این رو محققی به نام شواترز (Schwartz) و همکارانش در سال 1993، از روش امتیازدهی برای تشخیص بیماران استفاده کردند. این امتیازات براساس مشاهدات اکوکاردیوگرام، سابقه بالینی، تاریخچه خانوادگی و علایم بیمار، طراحی شده است (جدول 3). بر مبنای این سیستم امتیازدهی، احتمال LQTS مطابق رابطه زیر است: امتیاز کمتر از 1با احتمال LQTs کم، امتیاز بین 1/5 تا 3 با احتمال بروز متوسط و امتیاز بالاتر از 5 دارای احتمال بالای بروز LQTs میباشد.
شکل10: نقص کانال یونی در بروز LQT3 و اثر آن بر الکتروکاردیوگرام (24, 19)
شکل11: محرکهای آریتمیهای قلبی درLQT1 ، LQT2 و LQT3 (20)
جدول 2: تفکیک و تشخیص LQT1, LQT2, LQT3 از یکدیگر بر اساس جهشهای شایع (19)
جدول 3: مشاهدات الکتروکاردیوگرام و سیستم امتیاز دهی شوارتز و همکارانش (20)
رویکردهای درمانی بیماری LQTs: طبق مطالعات انجام شده محققان نشان دادهاند که درمان مبتنی بر RNAi میتواند در درمان بیماری LQTs موثر باشد. در جهشهای منفی غالب LQTs از RNAi برای خاموش کردن محصول پروتئینی استفاده میشود. لو و همکاران اثر RNAi را بر جهشهای منفی غالب LQT2 در موقعیت E637K در ژن hERG مورد بررسی قرار دادند. در این روش با استفاده از siRNA قسمت خاصی از توالی را در موقعیت E637K-hERG مورد هدف قرار دادند و عملکرد پروتئین را اصلاح کردند. این تکنیک اثرات جهش E637K-hERG را کاهش داد به طوریکه خواص کینتیکی پروتئین جهش یافته در حد سطوح پروتئین نرمال افزایش یافت (20,28). با این حال، مشخص است که اگر این فناوری در داخل بدن و در صورت کافی بودن یک کپی از پروتئین نرمال کار کند، پس وجود همان یک کپی از ژن برای حفظ عملکرد طبیعی در داخل بدن کافی است. یکی دیگر از روشهای درمانی، توانایی تولید سلول های بنیادی پرتوان القایی (iPSC)است که زمینه تحقیقات قلب و عروق را متحول کرده است، امکان تجزیه و تحلیل عملکردی بافت قلب بیمار در این روش فراهم میشود. فناوری سلولهای بنیادی به محققان این امکان را میدهد که یک نمونه خاص بیمار را از فیبروبلاستهای پوست از یک خانواده تولید کنند که در نهایت منجر به ایجاد جریان الکتریکی درون سلولی و بین سلولی در بیماران میشود (29). تکنیک دیگر، کاربرد CRISPR/Cas9 میباشد که یک تکنیک ویرایش ژنوم بسیار دقیق و کارآمد است که سریعتر و ارزانتر از سایر ویرایشهای ژنی قبلی است. این تکنیک میتواند خطوط جهش یافتههای ایزوژنیک را تولید کند به این صورت که با کنترلiPSCها یا ایجاد iPSCهای اصلاح شده ژنتیکی مربوط به نواحی جهش یافته میتوانند باعث از بین بردن تفاوت های اپی ژنتیکی یا ناشناخته شوند و تنوع فنوتیپی بیماری LQTs را اصلاح کنند. با اینحال تکنیکهای گفته شده دارای محدودیتهایی نیز هستند و چنانچه مورد نظر محققان باشند، لازم است اصلاحاتی در روشها صورت گیرد تا کارآیی آنها افزایش یابد (30). پیشرفتهای اخیر پیشنهاد میکنند که تکنیکهای RNAi, iPSC, CRISPR/Cas9 میتوانند امیدی برای درمان بیماریهای LQTs به شمار آیند (20).
سندرم QT کوتاه (SQT): سندرم SQT بیماری وخیم با الگوی توارثی اتوزوم غالب با نفوذ فنوتیپی بالا میباشد که در هر دو جنس و در تمام سنین با علائمی از جمله کوتاه شدن بازهQT ، فیبریلاسیون دهلیزی، آریتمی و تاکی کاردی بطنی همراه است که منجر به سنکوپ و مرگ ناگهانی قلبی میشود (31). دپولاریزاسیون سریع قلب موجب کاهش فاصلهQT بر روی ECG شده که زمینه ایجاد آریتمیهای قلبی که منجر به سنکوپ و مرگ ناگهانی میشود را بهوجود میآورد (34-32). بسیاری از مبتلایان در طول زندگی خود هرگز علائمی نداشتند و یا حتی اولین تجربه آنها، مرگ ناگهانی قلبی بوده است، به همین علت این بیماری مرگبار در اکثر موارد در آریتمیهای وخیم و همچنین نتایج مرگبار آن در جوانان تشخیص داده شده است (31,32).
اساس ژنتیکی و اهمیت کانالهای یونی: بیشتر از 30 جهش نادر در 8 ژن برای سندرم SQT که میتواند مادرزادی یا اکتسابی باشد، شناسایی شده است که بر حسب زیرگروههای SQTs ژنهای CACNA1C, CACNA2D1, CACNB2, KCNH2, KCNJ2, KCNQ1 مشخص شدهاند که در جدول4 آورده شده است (35,36):
با توجه به توضیحات جدول 4، جهش ها در مجموع، دو عملکرد مهم و حیاتی را شامل میشوند (35,36):
1-افزایش عملکرد پروتئین در ژنهای کدکننده کانالهای بیرون بر پتاسیم
2-کاهش عملکرد در زیرواحدهای متفاوت کانالهای کلسیم type-L قلبی و همچنین 2 ژن SLC4A3 و SCN5A که در جدول 5 بر حسب فنوتیپ با سایر ژنها آورده شده است (35): در جدول 5 دسته بندی جهشها آورده شده است که بهصورتزیر می توان آن را توضیح داد (35):
الف. جهشهای مرتبط با سندرم SQT
1- ژنهای کدکننده کانال پتاسیم KCNQ1, KCNj2, KCNH2
2- ژن SLC4A3 (35)
ب. جهشهای مرتبط با سندرم بروگادا که کوتاه شدن بازه QT را نشان می¬دهد، اما ثابت کنندهی تشخیص قطعی ابتلا به سندرم SQT نیست:
1- ژنهای کدکننده کانال کلسیم CACNB2, CACNA2D1, CACNA1C
2- ژن SCN5A (35,37)
همچنین جهش در 5 ژن دیگرKCNH2, KCNQ1, KCNJ2, CACNA1C, CACB2b نیز در افراد مبتلا یا مشکوک به سندرم SQT به دلیل کشنده بودن بررسی شده است (33). همانطور که در شکل 12 هم نشان داده شده است، ژنهای مختلفی که جهش در هر کدام از آنها منجر به بروز انواع آریتمی میشود، با هم همپوشانی دارند (33). اما چه تغییراتی در جریانهای یونی باعث کوتاه شدن بازه QT میشود؟ درواقع کاهش جریآنهای قطبش مجدد به سمت داخل (repolarizing inward)و یا همچنین افزایش جریانهای قطبش مجدد به سمت خارج (repolarizing outward ) باعث قطبش مجدد زودهنگام میشود. این قطبش مجدد زودهنگام، خود باعث کوتاه شدن دوره پتانسیل عمل و کوتاه شدن بازهQT می¬گردد. همانطور که در شکل 13 نشان داده شده است، جهشهای کسب عملکرد پتاسیم و جهشهای از دست دادن عملکرد کانالهای کلسیم و سدیم، منجر به یک فاز رپلاریزاسیون کوتاه شده در طول پتانسیل عمل و در نهایت منجر به کوتاه شدن فاصله QT میشود (33).
تشخیص SQTS: تشخیص سندرم SQTs بر اساس تاریخچه خانوادگی، نشانههای سنجش بیماری و نمودار الکتروکاردیوگرام بیمار انجام میشود (34,37). تشخیص بالینی بیماری با پیگیری حالتهای مختلف در بیمار انجام میگیرد که عبارتند از سابقه تپش قلب و سنکوپ، تاریخچه خانوادگی سنکوپ و مرگ ناگهانی یا بروز مرگ در سنین کم در خانواده. همچنین علتهای ثانویه بروز بیماری نیز باید مورد بررسی قرار گیرند، مانند: هیپرکلسمی، هیپرکالمی، تب شدید و اسیدوز. شوارتز و همکاران سیستم امتیازدهی خاصی را برای کمک به تشخیص سندرم SQTS پیشنهاد کردهاند که طبق جدول 6 دستهبندی می¬شود (37):
طبق جدول 6، امتیاز کمتر از 2 با احتمال کم بروز بیماری، امتیاز 3 با احتمال متوسط بروز بیماری و امتیاز بیشتر از 4 با احتمال بالای بیماری همراه است. اگر مقدار QTCحدود <300-320 ms باشد، طبق مطالعات جدید، بیمار دارای سندرم SQTS است و چنانچه مقدار QTC در محدوده<360 ms باشد در دسته SQTS4/SQTS5 قرار میگیرد. طبق مطالعه¬ای که ویسکین (Viskin) انجام داده است، زنان دارای QTC در محدوده <340 ms و مردان در محدوده <330 ms مبتلا به SQTS تشخیص داده میشوند. در خانوادههایی که دارای سابقه مرگهای ناگهانی قلبی هستند، این محدوده در زنان <370 ms و در مردان <360 ms میباشد (35,38). سندرم بروگادا: سندرم بروگادا (BrS) نوعی بیماری آریتموژنیک توارثی است که با اختلالات هدایت قلبی مشخص میشود و روی نوارهای اکوکاردیوگرام بهصورت بلندشدن قطعه ST (ST-segment elevation) دیده میشود (شکل14) (39,40). این بیماران شدیدا در معرض مرگ های ناگهانی قلبی (SCD) هستند که در نتیجه فیبریله شدن بطنی (Ventricular fibrillation) رخ میدهد (41). سندرم بروگادا عمدتاً در دوران بزرگسالی بروز میکند و متوسط سن مرگ ناگهانی تقریباً ۴۰ سال است. درواقع سندرم بروگادا، ناشی از اختلال کانال سدیم با اختلالات هدایت پیشرونده وابسته به سن، مانند طولانی شدن PQ ECG، QRS، و بازههای HV مرتبط است. اختلال در جریان سدیم به بلوکه شدن هدایت موضعی در اپیکاردیوم کمک میکند و منجر به چندین قله در کمپلکس QRS و تحریک فیبریلاسیون دهلیزی و بطنی میشود (39,42,43).
تاریخچه و ژنتیک سندرم بروگادا: در سال 1992 پدرو و جوزف بروگادا (Pedro and Josef Brugada)، برای اولین بار سندرم بروگادا را به عنوان یک بیماری توارثی معرفی کردند که باعث مرگ ناگهانی در خانوادهای میشد که دارای تاریخچه فامیلی از بروز چنین مرگهایی بودند (44,45). پس از آن در سال 1998 مشخص شد که جهشهای ژن SCN5A روی کروموزوم 3p21 با سندرم بروگادا ارتباط دارند. اگرچه تنها در 15-30% موارد جهشهای این ژن، مسئول بروز سندرم بروگادا شناخته شده است و در بقیه موارد، با وجود اینکه اغلب آنها خانوادگی هستند، ژن یا جایگاه کروموزومی خاصی تاکنون شناسایی نشده است (43,46). اولین ژن مرتبط به سندرم بروگادا، SCN5A است. جهشها در این ژن مسئول بروز LQT3 نیز هستند. تاکنون حدود 60 جهش در این ژن شناسایی شده است و تقریباً 24 جهش از میان آنها در سیستمهای بیانی مطالعه شدهاند و مشخص شده که اثر آنها بهصورت فقدان عملکرد (Loss of function) میباشد و به 4 حالت ممکن است عمل کنند (46):
1) باعث عدم بیان ژنهای کدکننده کانال سدیم میشوند.
2) کانالهای سدیم وابسته به ولتاژ و زمان هستند، یعنی در یک چرخه فعالشدن، غیرفعال شدن و دوباره فعالشدن، عمل میکنند. جهشها ممکن است این چرخه را دچار شیفت یا تغییر کنند.
3) جهشها باعث میشوند کانال سدیم وارد حالت حد واسط و غیرفعالی شود که خود سبب حرکت آهسته یونها میگردد.
4) جهشها گاه به طور خیلی سریع و برگشت ناپذیر، کانال سدیم را غیرفعال میکنند (40).
همانطور که در شکل 15 نشان داده شده است، رابطه بین جهشهای ژنی و کانالهای یونی و عملکرد آنها با سندرم بروگادا مشخص شده است (41).
جدول 4: زیر گروههای SQTS و ژنهای دخیل در بروز بیماری (35)
جدول5: ژنهای مرتبط با سندرم SQT و SQT interval
شکل 12: همپوشانی ژنهای مرتبط با LQTS, SQTS و سندرم بروگادا (33)
شکل13: بررسی جهشهای کسب عملکرد و از دست دادن عملکرد در کانالهای یونی و تاثیر در فاصله QT (33)
جدول6: امتیازدهی شوارتز برای تشخیص سندرم SQTS (35)
شکل14: دیاگرام ECG بیماران سندرم بروگادا. در این شکل بلند شدن قطعه ST در بیماران مشخص است (1)
شکل15: نمایش شماتیک یک کاردیومیوسیت که پروتئینهای دخیل در پاتوژنز سندرم بروگادا را نشان میدهد (41).
وراثت سندرم بروگادا: در بیشتر موارد، سندرم بروگادا به روش اتوزومال غالب با نفوذ ناقص به ارث میرسد، به استثنای سندرم بروگادایی که وابسته به KCNE5 است که به روش وابسته به X به ارث میرسد. درواقع بیشتر افرادی که مبتلا به سندرم بروگادا هستند، والدین مبتلا دارند که نسبت موارد ایجاد شده توسط یک واریانت بیماریزای جدید، 1% تخمین زده میشود. هر کودک از یک فرد مبتلا به سندرم بروگادای اتوزومال غالب، ۵۰% شانس به ارث بردن واریانت بیماریزا را دارد. تست پیش از تولد برای حاملگیهای پرخطر در صورتی امکانپذیر است که الل بیماری زا در خانواده شناخته شده باشد (41). اگر الل بیماریزا در DNA هیچ یک از والدین شناسایی نشود، ممکن است که یک الل بیماریزای جدید یا موزائیسم سلولهای جنسی در یک والد رخ داده باشد (البته تا به امروز، موزائیسم سلولهای جنسی در سندرم بروگادا توصیف نشده است). اگر چه بیشتر افرادی که مبتلا به سندرم بروگادا تشخیص داده میشوند، الل بیماریزا را از یک والد به ارث بردهاند، تاریخچه خانواده ممکن است به دلیل عدم تشخیص اختلال در اعضای خانواده، نفوذ ناقص، مرگ زودهنگام والدین قبل از شروع علائم، یا شروع دیر هنگام علائم در والدین مبتلا، منفی به نظر برسد (47-42). تشخیص براساس یافتههای بالینی و یا به وسیله شناسایی یک واریانت اللی بیماری¬زای هتروزیگوت (یا هموزیگوت در5 KCNE) در یکی از ۲۳ ژن زیر است:ABCC9, CACNA1C, CACNA2D1, CACNB2, FGF12, GPD1L, HCN4, KCND2, KCND3, KCNE5, KCNE3, KCNH2, KCNJ8, PKP2, RANGRF, SCN1B, SCN2B, SCN3B, SCN5A, SCN10A, SEMA3A, SLMAP, and TRPM4. (42). مطالعات مختلف نشان میدهند که هیچ فنوتیپ دیگری با واریانت بیماریزای ایجاد کننده سندرم بروگادا که در GPDL1،HCN4 و RANGR شناسایی شدهاند، مرتبط نیست. تعدادی از این ژنها در (جدول7) آورده شده است (41):
جدول7: ژن¬های مرتبط با سندرم بروگادا (41)
SCN5A: رابطه بین ژن بیماری¬زای SCN5A و سندرم بروگادا در سال ۱۹۹۸ شناسایی شد، ژن SCN5A به عنوان اولین ژن دخیل در سندرم بروگادا شناسایی شد که متداولترین ژن مرتبط با این سندرم نیز معرفی شده است، با این وجود، بیش از 10 سال است که ژن¬های مرتبط با این سندرم در حال شناسایی هستند (44). تاکنون بیش از ۱۰۰ نوع جهش بیماریزای مختلف در SCN5A گزارش شدهاند که تقریبا نیمی از آنها از نظر بالینی تایید شدهاند (46). چندین نوع جهش بیماریزای مختلف که بر ساختار، عملکرد و عبور از کانالهای سدیم تاثیر میگذارند نیز شناسایی شدهاند (42). توالی ژنومی این ژن، شامل بیش از ۱۰۰ کیلوباز طول دارد. این ژن شامل ۲۸ اگزون است. ژن SCN5A زیر واحد آلفا کانال سدیم قلب را رمزگذاری میکند و مسئول فاز صفر پتانسیل عمل قلب است. پروتئین آن حاوی ۲۰۱۶ اسید آمینه است که شامل چهار تکرار داخلی است، هر کدام از آنها دارای پنج بخش هیدروفوبیک (S۱، S۲، S۳، S۵، S۶) و یک بخش دارای بار مثبت (S۴) میباشند. بخش S۴ احتمالاً سنسور ولتاژ است و با یک سری اسید آمینه با بار مثبت مشخص میشود (48). این پروتئین غشایی یکپارچه، نفوذپذیری یون سدیم وابسته به ولتاژ را در غشاهای تحریکپذیر تعدیل میکند. با فرض اینکه ترکیبات باز یا بسته نسبت به اختلاف ولتاژ در طول غشا واکنش نشاندهند، پروتئین یک کانال انتخابی سدیم را تشکیل میدهد که ممکن است یونهایNa+ را مطابق با گرادیان الکتروشیمیایی عبور دهند. این کانال مسئول افزایش اولیه پتانسیل عمل در ECG است. این پروتئین در ماهیچه قلب دهلیز و بطن انسان بیان میشود، اما در ماهیچه اسکلتی، مغز، میومتر، کبد و یا طحال وجود ندارد (42,48) (شکل16). انواع پاتوژنیک و موتانت در ژن SCN5A منجر به کاهش جریانNa+ توسط یکی از دو مکانیزم اصلی در سلول میشود: عدم بیان کافی از کانال تغییر یافته یا غیرفعال شدن سریع کانال که هر دو حالت منجر به تغییر در پتانسیل عمل سلولهای قلب میگردد که در نهایت منجر به آریتمی میشود (46,42).
نقش کانالهای یونی در بروگادا: چون ژن SCN5A کدکننده زیر واحد α کانال سدیم قلب است و جهش در آن باعث کاهش جریان سدیم دپولاریزه کننده و بروز علایم سندرم بروگادا میشود، این سندرم را به عنوان نوعی بیماری کانال یونی (Channelopathies disease) میشناسند (46). در سال 1995 مشخص شد، ژن SCN5A مسئول بروز گروهی از بیماران LQTS (LQT3) نیز هستند که گروه نسبتاً نادری از LQTSها را شامل میشود. جهشهای این ژن در LQTS، باعث غیرفعال شدن جریان سدیم (یک جریان دپولاریزهکننده) در طی فاز پلاتو (Plateau phase) پتانسیل عمل، یا به عبارت دیگر، کسب عملکرد (Gain of function) زیاد کانال میشود که منجر به طولانی شدن رپلاریزاسیون و بنابراین حالتLong QT میشود (46). دیگر جهشهای این ژن باعث بروز علایم بالینی متفاوتی مانند بالارفتن موج ST در سندرم بروگادا میشود، اما در سندرم بروگادا، جهشهای اخیر سبب فقدان عملکرد کانال میگردد. بنابراین میتوان گفت جهشهای ژن SCN5A منجر به بروز فنوتیپهای کلینیکی هتروژنوس و متفاوتی میشود. اما در همه بیماریهای ایجاد شده توسط این جهشها (LQTS، سندرم بروگادا، بیماری غده سینوسی و بیماریهای انتقال یونی) مرگ ناگهانی، غالباً در خواب و استراحت رخ میدهند که محققان دلیل آن را به هم خوردن تعادل و هماهنگی بین ایمپالسهای سمپاتیک و پاراسمپاتیک میدانند که مهمترین عامل بروز آریتمی است (شکل16) (46).
میزان شیوع سندرم مرگ شبانه: به دلیل کم بودن اطلاعات ژنتیکی موجود در این سندرم، میزان نفوذ بیماری در میان جمعیتها، هنوز به طور دقیق تعیین نشده است. تخمین محققان 10000/ 5-1 نفر در کشورهای غربی است. اما شیوع بالای سندرم (2500/1) در کشورهای شرقی بهویژه تایلند نیز دیده میشود. در جنوب آسیا مانند فیلیپین، ژاپن و تایلند، چون این سندرم باعث مرگ در حالت خواب و در نیمههای شب میشود، از این سندرم به عنوان مرگ شبانه بدون توضیح و ناگهانی (Sudden Unexplained Nocturnal Death) یاد میکنند که بیشتر در افراد جوان رخ میدهد (40). توارث سندرم بروگادا، اتوزومال غالب است و بیشتر بهصورت تاکی کاردیای بطنی چند شکلی (Polymorphic ventricular tachycardia) و مرگ ناگهانی در بیماران جوان و در درجه کمتر در نوزادان و کودکانی دیده میشود که دارای قلبهای سالم از نظر ساختاری هستند. اگرچه میانگین سنی در زمان اولین تشخیص یا مرگ ناگهانی قلبی، حدود 40 سالگی است، اما فنوتیپ بروگادا در محدوده سنی وسیعی گزارش شده است. این سندرم مسئول 20% از مرگهای ناگهانی در بیمارانی با قلبهای سالم است (40). در اکوکاردیوگرام این بیماران علاوه بر بلندشدن قطعه ST، طولانی شدن فاصله QT هم دیده میشود. با این وجود در برخی از بیماران الگوی اکوکاردیوگرام هم در تشخیص بیماری ناتوان است (49). مشخصات کلی این سندرم در جدول 8 ذکر شده است. در LQTs، سن وقوع اولین علایم بیماری، دهههای اولیه زندگی است، ولی در سندرم بروگادا، این علایم در دهه چهارم حیات فرد بیشتر دیده میشود. مشخص شده است که سندرمهای LQTs و بروگادا دارای نقصهای ژنتیکی مشابه هستند، اما علایم کلینیکی وابسته به سن متفاوتی را نشان میدهند. دلیل این گونه بروزهای متفاوت هنوز مشخص نشده است، ولی احتمال دارد به این دلیل باشد که این سندرمها بهصورت مولتی فاکتوریال یا چند عاملی هستند (49). از میان بیماران گزارش شده با سندرم بروگادا، حدود 90% آنها مرد بودند و میانگین سنی
آنها بین 22 تا 65 سال گزارش شده است. محققین با مشاهده بروز زودتر علایم LQTs و بروگادا سندرم، در مردان پیشنهاد میکنند که جنسیت و فعالیت های فیزیکی در مردان در ظهور علایم کلینیکی نقش دارد (49). از فاکتورهای دیگری که در بروز علایم ناگهانی این دو سندرم نقش دارند: تغییرات هورمونی، اختلالات الکترولیتی، داروهای مصرفی بیمار، میزان جذب غذا، تب شدید و سن را می توان نام برد. هیچ کدام از این فاکتورها مستقیما در افزایش وخامت علایم بالینی نقش ندارند و بنابراین بهتر است از واژه تعدیل کننده (Modifier) برای آنها استفاده شود. با این وجود جهشهای شناسایی شده، تنها 30 تا 40% از موارد سندرم را توجیه میکنند و تاکنون بقیه عوامل دخیل در بروز سندرم بروگادا ناشناخته مانده است. از این رو یکی از احتمالات محققین، دخیل بودن ژنوم میتوکندری و وجود اختلالات زنجیره تنفسی، در بروز این سندرم است (48,50).
شکل16: بررسی کانال سدیمی SCN5A و عملکرد دومین پروتئینی در بروز سندرم بروگادا (48)
جدول 8: متغیرهای شناسایی شده مرتبط با سندرم بروگادا و مرگ ناگهانی قلبی(SCD) (49)
نتیجهگیری
درک فعلی ما از پاتوفیزیولوژی اختلالات آریتموژنیک قلبی در 20 سال گذشته، عمدتاً از تحقیقات گسترده در مورد کانالوپاتیهای یونی، به ویژه LQTS مادرزادی و سایر موارد مانند سندرم بروگادا به دست آمده است. درک بهتر محققین از عملکرد هر کانال یونی در رابطه با جهشهای ژنی، و نحوه اثر هر تغییر ژنتیکی در عملکرد کانالهای یونی قلب، راه را برای تحقیق و توسعه روشهای درمانی هدفمند دارویی و غیردارویی مؤثرتر، مانند ژن درمانی در بیماران هموار کرده است.
حامی مالی: ندارد.
تعارض در منافع: وجود ندارد.
References:
1- Shah SR, Park K, Alweis R. vLong Qt Syndrome: A Comprehensive Review of the Literature and Current Evidence. Current Problems in Cardiology 2019; 44(3): 92-106.
2- Dupre A, Vincent S, Iaizzo PA. Basic ECG Theory, Recordings, and Interpretation. In: Handbook of Cardiac Anatomy, Physiology, and Devices: Springer; 2005: 191-201.
3- Gigli M, Merlo M, Graw SL, Barbati G, Rowland TJ, Slavov DB, et al. Genetic Risk of Arrhythmic Phenotypes in Patients with Dilated Cardiomyopathy. J American College of Cardiology 2019; 74(11): 1480-90.
4- Lei M, Wu L, Terrar DA, Huang CL-H. Modernized Classification of Cardiac Antiarrhythmic Drugs. Circulation 2018; 138(17): 1879-96.
5- Shaffer F, Mccraty R, Zerr CL. A Healthy Heart is Not a Metronome: An Integrative Review of the Heart's Anatomy and Heart Rate Variability. Front Psychol 2014; 5: 1040.
6- Bezzina CR, Lahrouchi N, Priori SG. Genetics of Sudden Cardiac Death. Circul Res 2015; 116(12): 1919-36.
7- Bernardi J, Aromolaran KA, Zhu H, Aromolaran AS. Circadian Mechanisms: Cardiac in Channel Remodeling and Arrhythmias. Front Physiol 2021; 11: 611860.
8- Moreau A, Gosselin-Badaroudine P, Mercier A, Burger B, Keller DI, Chahine M. A Leaky Voltage Sensor Domain of Cardiac Sodium Channels Causes Arrhythmias Associated with Dilated Cardiomyopathy. Scientific Reports 2018; 8(1): 1-12.
9- Lazzerini PE, Laghi-Pasini F, Boutjdir M, Capecchi PL. Cardioimmunology of Arrhythmias: The Role of Autoimmune and Inflammatory Cardiac Channelopathies. Nature Reviews Immunology 2019; 19(1): 63-4.
10- Knollmann BC, Roden DM. A Genetic Framework for Improving Arrhythmia Therapy. Nature 2008; 451(7181): 929-36.
11- Giudicessi JR, Wilde AA, Ackerman MJ. The Genetic Architecture of Long QT Syndrome: A Critical Reappraisal. Trends in Cardiovascular Medicine 2018; 28(7): 453-64.
12- Shomanova Z, Ohnewein B, Schernthaner C, Höfer K, Pogoda CA, Frommeyer G, et al. Classic and Novel Biomarkers as Potential Predictors of Ventricular Arrhythmias and Sudden Cardiac Death. J Clinical Medicine 2020; 9(2): 578.
13- Marian AJ, Asatryan B, Wehrens XH. Genetic Basis and Molecular Biology of Cardiac Arrhythmias in Cardiomyopathies, Cardiovascular Research 2020; 116(9): 1600-19.
14- Hammerer-Lercher A, Namdar M, Vuilleumier N. Emerging Biomarkers for Cardiac Arrhythmias. Clinical Biochemistry 2020; 751-6: 1-6.
15- Khatami M, Houshmand M, Sadeghizadeh M, Eftekharzadeh M, Heidari MM, Saber S, et al. Accumulation of Mitochondrial Genome Variations in Persian Lqts Patients: A Possible Risk Factor?, Cardiovasc Pathology 2010; 19(2): E21-E7.
16- Group JJW. Guidelines for Risks and Prevention of Sudden Cardiac Death (JCS 2010)–Digest Version. Cir J 2012; 76(2): 489-507.
17- Landstrom AP, Dobrev D, Wehrens XH. Calcium Signaling and Cardiac Arrhythmias. Cir Res 2017; 120(12): 1969-93.
18- Ponce-Balbuena D, Deschênes I. Long Qt Syndrome–Bench to Bedside. Heart Rhythm O2 2021 J; 2(1): 89-106.
19- Wallace E, Howard L, Liu M, O’Brien T, Ward D, Shen S, et al. Long Qt Syndrome: Genetics and Future Perspective. Pediat Cardiol 2019; 40(7): 1419-30.
20- Hintsa T, Puttonen S, Toivonen L, Kontula K, Swan H, Keltikangas-Järvinen L. A History of Stressful Life Events, Prolonged Mental Stress and Arrhythmic Events in Inherited Long Qt Syndrome. Heart 2010; 96(16): 1281-6.
21- Rivaud MR, Delmar M, Remme CA. Heritable Arrhythmia Syndromes Associated with Abnormal Cardiac Sodium Channel Function: Ionic and Non-Ionic Mechanisms. Cardiovasc Res 2020; 116(9): 1557-70.
22- Khatami M, Heidari M. Identification of a Large-Scale Mitochondrial Dna Deletion in Iranian Heart Arrhythmia Patients(Lqt Syndrome). JSSU 2011; 18(6): 531-9. [Persian]
23- Bohnen M, Peng G, Robey S, Terrenoire C, Iyer V, Sampson K, Kass R. Molecular Pathophysiology of Congenital Long Qt Syndrome. Physiol Rev 2017; 97(1): 89-134.
24- Tester DJ, Ackerman MJ. Genetics of Long Qt Syndrome. Methodist Debakey Cardiovascular Journal 2014; 10(1): 29-33.
25- Booker P, Whyte S, Ladusans E. Long Qt Syndrome and Anaesthesia. Br J Anaesth 2003; 90(3): 349-66.
26- Khatami M, Eghbali Ebrahimabadi M. Molecular Analysis of Trnaglu, Trp, Ala, Asp Genes of Mtdna in Patients with Long Qt Syndrome. J Mazandaran Univ Med Sci 2014; 24(116): 141-8. [Persian]
27- Li G, Ma S, Sun C. Rna Interference-Based Therapeutics for Inherited Long Qt Syndrome. Exp Ther Med 2015; 10(2): 395-400.
28- Sala L, Gnecchi M, Schwartz PJ. Long Qt Syndrome Modelling with Cardiomyocytes Derived from Human-Induced Pluripotent Stem Cells. Arrhythm & Electrophysiol Rev 2019; 8(2): 105-10.
29- Song Y, Zheng Z, Lian J. Deciphering Common Long Qt Syndrome Using Crispr/Cas9 in Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Front in Cardiovasc Med 2022; 9: 889519.
30- Schimpf R, Wolpert C, Gaita F, Giustetto C, Borggrefe M. Short QT Syndrome. Cardiovas Res 2005; 67(3): 357-66.
31- Mazzanti A, Maragna R, Vacanti G, Kostopoulou A, Marino M, Monteforte N, et al. Hydroquinidine Prevents Life-Threatening Arrhythmic Events in Patients with Short Qt Syndrome. J American College of Cardiology 2017; 70(24): 3010-5.
32- Campuzano O, Sarquella-Brugada G, Cesar S, Arbelo E, Brugada J, Brugada R. Recent Advances in Short Qt Syndrome. Front Cardiovasc Med 2018; 5: 149.
33- Walsh R, Adler A, Amin AS, Abiusi E, Care M, Bikker H, et al. Evaluation of Gene Validity for Cpvt and Short Qt Syndrome in Sudden Arrhythmic Death. Eur Heart J 2022; 43(15): 1500-10.
34- Dewi IP, Dharmadjati BB. Short Qt Syndrome: The Current Evidences of Diagnosis and Management. J Arrhythmia 2020; 36(6): 962-6.
35- Raschwitz LS, El-Battrawy I, Schlentrich K, Besler J, Veith M, Roterberg G, et al. Differences in Short Qt Syndrome Subtypes: A Systematic Literature Review and Pooled Analysis. Front Genet 2020; 10: 1312.
36- Campuzano O, Fernandez-Falgueras A, Lemus X, Sarquella-Brugada G, Cesar S, Coll M, et al. Short Qt Syndrome: A Comprehensive Genetic Interpretation and Clinical Translation of Rare Variants. J Clin Med 2019; 8(7): 1035.
37- Viskin S. The Qt Interval: Too Long, Too Short or Just Right. Heart Rhythm 2009; 6(5): 711-5.
38- Ciconte G, Monasky MM, Santinelli V, Micaglio E, Vicedomini G, Anastasia L, et al. Brugada Syndrome Genetics is Associated with Phenotype Severity. Eur Heart J 2021; 42(11): 1082-90.
39- Pappone C, Santinelli V. Brugada Syndrome: Progress in Diagnosis and Management. Arrhythm & Electrophysiol Rev 2019; 8(1): 13-18.
40- Juang J-MJ, Horie M. Genetics of Brugada Syndrome. J Arrhythmia 2016; 32(5): 418-25.
41- Brugada R, Campuzano O, Sarquella-Brugada G, Brugada P, Brugada J, Hong K. Brugada Syndrome. Methodist Debakey Cardiovasc J 2014; 10(1): 25-8.
42- Sieira J, Brugada P. The Definition of the Brugada Syndrome. Eur Heart J 2017; 38(40): 3029-34.
43- Coppola G, Corrado E, Curnis A, Maglia G, Oriente D, Mignano A, Brugada P. Update on Brugada Syndrome 2019. Curr Probl Cardiol 2021; 46(3): 100454.
44- Moradimoghadam O, Sedaghat A, Niakan M, Hajiesmaeili M, Seifi S, Soleimanirad R. Sudden Cardiac Arrest Due to Brugada Syndrome: A Case Report and Literature Review. JSSU 2013; 21(1): 113-7. [Persian]
45- Wilde AA, Amin AS. Clinical Spectrum of SCN5A Mutations: Long QT Syndrome, Brugada Syndrome, and Cardiomyopathy. JACC: Clin Electrophysiol 2018; 4(5): 569-79.
46- Michowitz Y, Milman A, Andorin A, Sarquella-Brugada G, Gonzalez Corcia MC, Gourraud J-B, et al. Characterization and Management of Arrhythmic Events in Young Patients with Brugada Syndrome. J Am Coll Cardiol 2019; 73(14): 1756-65.
47- Antzelevitch C. Brugada Syndrome. Pacing and Clinical Electrophysiology 2006; 29(10): 1130-59.
48- Gourraud J-B, Barc J, Thollet A, Le Marec H, Probst V. Brugada Syndrome: Diagnosis, Risk Stratification and Management. Archives of Cardiovascular Diseases 2017; 110(3): 188-95.
49- Fallah Tafti M, Khatami M, Rezaei S, Heidari MM, Hadadzadeh M. Novel and Heteroplasmic Mutations in Mitochondrial Trna Genes in Brugada Syndrome. Cardiol J 2018; 25(1): 113-19.
1- Shah SR, Park K, Alweis R. vLong Qt Syndrome: A Comprehensive Review of the Literature and Current Evidence. Current Problems in Cardiology 2019; 44(3): 92-106.
2- Dupre A, Vincent S, Iaizzo PA. Basic ECG Theory, Recordings, and Interpretation. In: Handbook of Cardiac Anatomy, Physiology, and Devices: Springer; 2005: 191-201.
3- Gigli M, Merlo M, Graw SL, Barbati G, Rowland TJ, Slavov DB, et al. Genetic Risk of Arrhythmic Phenotypes in Patients with Dilated Cardiomyopathy. J American College of Cardiology 2019; 74(11): 1480-90.
4- Lei M, Wu L, Terrar DA, Huang CL-H. Modernized Classification of Cardiac Antiarrhythmic Drugs. Circulation 2018; 138(17): 1879-96.
5- Shaffer F, Mccraty R, Zerr CL. A Healthy Heart is Not a Metronome: An Integrative Review of the Heart's Anatomy and Heart Rate Variability. Front Psychol 2014; 5: 1040.
6- Bezzina CR, Lahrouchi N, Priori SG. Genetics of Sudden Cardiac Death. Circul Res 2015; 116(12): 1919-36.
7- Bernardi J, Aromolaran KA, Zhu H, Aromolaran AS. Circadian Mechanisms: Cardiac in Channel Remodeling and Arrhythmias. Front Physiol 2021; 11: 611860.
8- Moreau A, Gosselin-Badaroudine P, Mercier A, Burger B, Keller DI, Chahine M. A Leaky Voltage Sensor Domain of Cardiac Sodium Channels Causes Arrhythmias Associated with Dilated Cardiomyopathy. Scientific Reports 2018; 8(1): 1-12.
9- Lazzerini PE, Laghi-Pasini F, Boutjdir M, Capecchi PL. Cardioimmunology of Arrhythmias: The Role of Autoimmune and Inflammatory Cardiac Channelopathies. Nature Reviews Immunology 2019; 19(1): 63-4.
10- Knollmann BC, Roden DM. A Genetic Framework for Improving Arrhythmia Therapy. Nature 2008; 451(7181): 929-36.
11- Giudicessi JR, Wilde AA, Ackerman MJ. The Genetic Architecture of Long QT Syndrome: A Critical Reappraisal. Trends in Cardiovascular Medicine 2018; 28(7): 453-64.
12- Shomanova Z, Ohnewein B, Schernthaner C, Höfer K, Pogoda CA, Frommeyer G, et al. Classic and Novel Biomarkers as Potential Predictors of Ventricular Arrhythmias and Sudden Cardiac Death. J Clinical Medicine 2020; 9(2): 578.
13- Marian AJ, Asatryan B, Wehrens XH. Genetic Basis and Molecular Biology of Cardiac Arrhythmias in Cardiomyopathies, Cardiovascular Research 2020; 116(9): 1600-19.
14- Hammerer-Lercher A, Namdar M, Vuilleumier N. Emerging Biomarkers for Cardiac Arrhythmias. Clinical Biochemistry 2020; 751-6: 1-6.
15- Khatami M, Houshmand M, Sadeghizadeh M, Eftekharzadeh M, Heidari MM, Saber S, et al. Accumulation of Mitochondrial Genome Variations in Persian Lqts Patients: A Possible Risk Factor?, Cardiovasc Pathology 2010; 19(2): E21-E7.
16- Group JJW. Guidelines for Risks and Prevention of Sudden Cardiac Death (JCS 2010)–Digest Version. Cir J 2012; 76(2): 489-507.
17- Landstrom AP, Dobrev D, Wehrens XH. Calcium Signaling and Cardiac Arrhythmias. Cir Res 2017; 120(12): 1969-93.
18- Ponce-Balbuena D, Deschênes I. Long Qt Syndrome–Bench to Bedside. Heart Rhythm O2 2021 J; 2(1): 89-106.
19- Wallace E, Howard L, Liu M, O’Brien T, Ward D, Shen S, et al. Long Qt Syndrome: Genetics and Future Perspective. Pediat Cardiol 2019; 40(7): 1419-30.
20- Hintsa T, Puttonen S, Toivonen L, Kontula K, Swan H, Keltikangas-Järvinen L. A History of Stressful Life Events, Prolonged Mental Stress and Arrhythmic Events in Inherited Long Qt Syndrome. Heart 2010; 96(16): 1281-6.
21- Rivaud MR, Delmar M, Remme CA. Heritable Arrhythmia Syndromes Associated with Abnormal Cardiac Sodium Channel Function: Ionic and Non-Ionic Mechanisms. Cardiovasc Res 2020; 116(9): 1557-70.
22- Khatami M, Heidari M. Identification of a Large-Scale Mitochondrial Dna Deletion in Iranian Heart Arrhythmia Patients(Lqt Syndrome). JSSU 2011; 18(6): 531-9. [Persian]
23- Bohnen M, Peng G, Robey S, Terrenoire C, Iyer V, Sampson K, Kass R. Molecular Pathophysiology of Congenital Long Qt Syndrome. Physiol Rev 2017; 97(1): 89-134.
24- Tester DJ, Ackerman MJ. Genetics of Long Qt Syndrome. Methodist Debakey Cardiovascular Journal 2014; 10(1): 29-33.
25- Booker P, Whyte S, Ladusans E. Long Qt Syndrome and Anaesthesia. Br J Anaesth 2003; 90(3): 349-66.
26- Khatami M, Eghbali Ebrahimabadi M. Molecular Analysis of Trnaglu, Trp, Ala, Asp Genes of Mtdna in Patients with Long Qt Syndrome. J Mazandaran Univ Med Sci 2014; 24(116): 141-8. [Persian]
27- Li G, Ma S, Sun C. Rna Interference-Based Therapeutics for Inherited Long Qt Syndrome. Exp Ther Med 2015; 10(2): 395-400.
28- Sala L, Gnecchi M, Schwartz PJ. Long Qt Syndrome Modelling with Cardiomyocytes Derived from Human-Induced Pluripotent Stem Cells. Arrhythm & Electrophysiol Rev 2019; 8(2): 105-10.
29- Song Y, Zheng Z, Lian J. Deciphering Common Long Qt Syndrome Using Crispr/Cas9 in Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Front in Cardiovasc Med 2022; 9: 889519.
30- Schimpf R, Wolpert C, Gaita F, Giustetto C, Borggrefe M. Short QT Syndrome. Cardiovas Res 2005; 67(3): 357-66.
31- Mazzanti A, Maragna R, Vacanti G, Kostopoulou A, Marino M, Monteforte N, et al. Hydroquinidine Prevents Life-Threatening Arrhythmic Events in Patients with Short Qt Syndrome. J American College of Cardiology 2017; 70(24): 3010-5.
32- Campuzano O, Sarquella-Brugada G, Cesar S, Arbelo E, Brugada J, Brugada R. Recent Advances in Short Qt Syndrome. Front Cardiovasc Med 2018; 5: 149.
33- Walsh R, Adler A, Amin AS, Abiusi E, Care M, Bikker H, et al. Evaluation of Gene Validity for Cpvt and Short Qt Syndrome in Sudden Arrhythmic Death. Eur Heart J 2022; 43(15): 1500-10.
34- Dewi IP, Dharmadjati BB. Short Qt Syndrome: The Current Evidences of Diagnosis and Management. J Arrhythmia 2020; 36(6): 962-6.
35- Raschwitz LS, El-Battrawy I, Schlentrich K, Besler J, Veith M, Roterberg G, et al. Differences in Short Qt Syndrome Subtypes: A Systematic Literature Review and Pooled Analysis. Front Genet 2020; 10: 1312.
36- Campuzano O, Fernandez-Falgueras A, Lemus X, Sarquella-Brugada G, Cesar S, Coll M, et al. Short Qt Syndrome: A Comprehensive Genetic Interpretation and Clinical Translation of Rare Variants. J Clin Med 2019; 8(7): 1035.
37- Viskin S. The Qt Interval: Too Long, Too Short or Just Right. Heart Rhythm 2009; 6(5): 711-5.
38- Ciconte G, Monasky MM, Santinelli V, Micaglio E, Vicedomini G, Anastasia L, et al. Brugada Syndrome Genetics is Associated with Phenotype Severity. Eur Heart J 2021; 42(11): 1082-90.
39- Pappone C, Santinelli V. Brugada Syndrome: Progress in Diagnosis and Management. Arrhythm & Electrophysiol Rev 2019; 8(1): 13-18.
40- Juang J-MJ, Horie M. Genetics of Brugada Syndrome. J Arrhythmia 2016; 32(5): 418-25.
41- Brugada R, Campuzano O, Sarquella-Brugada G, Brugada P, Brugada J, Hong K. Brugada Syndrome. Methodist Debakey Cardiovasc J 2014; 10(1): 25-8.
42- Sieira J, Brugada P. The Definition of the Brugada Syndrome. Eur Heart J 2017; 38(40): 3029-34.
43- Coppola G, Corrado E, Curnis A, Maglia G, Oriente D, Mignano A, Brugada P. Update on Brugada Syndrome 2019. Curr Probl Cardiol 2021; 46(3): 100454.
44- Moradimoghadam O, Sedaghat A, Niakan M, Hajiesmaeili M, Seifi S, Soleimanirad R. Sudden Cardiac Arrest Due to Brugada Syndrome: A Case Report and Literature Review. JSSU 2013; 21(1): 113-7. [Persian]
45- Wilde AA, Amin AS. Clinical Spectrum of SCN5A Mutations: Long QT Syndrome, Brugada Syndrome, and Cardiomyopathy. JACC: Clin Electrophysiol 2018; 4(5): 569-79.
46- Michowitz Y, Milman A, Andorin A, Sarquella-Brugada G, Gonzalez Corcia MC, Gourraud J-B, et al. Characterization and Management of Arrhythmic Events in Young Patients with Brugada Syndrome. J Am Coll Cardiol 2019; 73(14): 1756-65.
47- Antzelevitch C. Brugada Syndrome. Pacing and Clinical Electrophysiology 2006; 29(10): 1130-59.
48- Gourraud J-B, Barc J, Thollet A, Le Marec H, Probst V. Brugada Syndrome: Diagnosis, Risk Stratification and Management. Archives of Cardiovascular Diseases 2017; 110(3): 188-95.
49- Fallah Tafti M, Khatami M, Rezaei S, Heidari MM, Hadadzadeh M. Novel and Heteroplasmic Mutations in Mitochondrial Trna Genes in Brugada Syndrome. Cardiol J 2018; 25(1): 113-19.
ارسال پیام به نویسنده مسئول
| بازنشر اطلاعات | |
 |
این مقاله تحت شرایط Creative Commons Attribution-NonCommercial 4.0 International License قابل بازنشر است. |
