

دوره 31، شماره 1 - ( فروردین 1402 )
جلد 31 شماره 1 صفحات 6312-6301 |
برگشت به فهرست نسخه ها


![]()
![]()
![]()
Download citation:
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:



BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
Hosseini Askarabadi S, Kaki A. Effect of Aerobic Exercise on IL-35 Anti-Inflammatory Cytokine and Behavioral Pain Responses in a Model of Diabetic Neuropathy Rats. JSSU 2023; 31 (1) :6301-6312
URL: http://jssu.ssu.ac.ir/article-1-5756-fa.html
URL: http://jssu.ssu.ac.ir/article-1-5756-fa.html
حسینی عسکرآبادی سیروس، کاکی احمد. بررسی اثر تمرین هوازی بر سایتوکین ضدالتهابی اینترلوکین ۳۵ و پاسخهای رفتاری درد در مدل موش صحرایی نوروپاتی دیابت. مجله علمي پژوهشي دانشگاه علوم پزشكي شهید صدوقی يزد. 1402; 31 (1) :6301-6312



متن کامل [PDF 951 kb]
(600 دریافت)
| چکیده (HTML) (1837 مشاهده)
References:
1- Feldman EL, Nave KA, Jensen TS, Bennett DL. New Horizons in Diabetic Neuropathy: Mechanisms, Bioenergetics, and Pain. Neuron 2017; 93(6): 1296-313.
2- Calcutt NA. Diabetic Neuropathy and Neuropathic Pain: A (Con) Fusion of Pathogenic Mechanisms? Pain 2020; 161(Suppl 1): S65-8.
3- Schreiber AK, Nones CF, Reis RC, Chichorro JG, Cunha JM. Diabetic Neuropathic Pain: Physiopathology and Treatment. World J of diabetes 2015; 6(3): 432-44.
4- Sai Laxmi M, Prabhakar O. Inflammatory Biomarkers as a Part of Diagnosis in Diabetic Peripheral Neuropathy. Journal of Diabetes and Metabolic Disorders 2021; 20: 869-82.
5- Pop-Busui R, Ang L, Holmes C, Gallagher K, Feldman EL. Inflammation as a Therapeutic Target for Diabetic Neuropathies. Curr Diab Rep 2016; 16(3): 29.
6- Li X, Mai J, Virtue A, Yin Y, Gong R, Sha X, et al. IL-35 is a Novel Responsive Anti-Inflammatory Cytokine—a New System of Categorizing Anti-Inflammatory Cytokines. PloS One 2012; 7(3): e33628.
7- Yang XF, Li X, Mai J, Virtue A, Yin Y, Sha X, et al. IL-35 is a Novel Responsive Anti-Inflammatory Cytokine-A New System of Categorizing Anti-Inflammatory Cytokines. American Society of Hematology; 2012; 120(21): 5197.
8- Jiang Y, Wang J, Li H, Xia L. IL-35 Alleviates Inflammation Progression in a Rat Model of Diabetic Neuropathic Pain Via Inhibition of JNK Signaling. J Inflamm (Lond) 2019; 16: 19.
9- Jiang Y, Wang J, Li H, Xia L. IL-35 Promotes Microglial M2 Polarization in a Rat Model of Diabetic Neuropathic Pain. Arch Biochem Biophys 2020; 685: 108330.
10- Hosseini A, Abdollahi M. Diabetic Neuropathy and Oxidative Stress: Therapeutic Perspectives. Oxid Med Cell Longev 2013; 2013: 168039.
11- Singleton JR, Smith AG, Marcus RL. Exercise as Therapy for Diabetic and Prediabetic Neuropathy. Curr Diab Rep 2015; 15(12): 120.
12- Yan J-E, Yuan W, Lou X, Zhu T. Streptozotocin-Induced Diabetic Hyperalgesia in Rats is Associated with Upregulation of Toll-Like Receptor 4 Expression. Neurosci lett 2012; 526(1): 54-8.
13- Morrow TJ. Animal Models of Painful Diabetic Neuropathy: the STZ Rat Model. Current Protocols in Neuroscience 2004; Chapter 9: Unit 9.18.
14- Wei M, Ong L, Smith MT, Ross FB, Schmid K, Hoey AJ, et al. The Streptozotocin‐Diabetic Rat as a Model of the Chronic Complications of Human Diabetes. Heart Lung Circ 2003; 12(1): 44-50.
15- Malmberg AB, Bannon AW. Models of Nociception: Hot‐Plate, Tail‐Flick, and Formalin Tests in Rodents. Current Protocols in Neuroscience 1999; 6(1): 8.9. 1-8.9. 15.
16- Chen YW, Hsieh PL, Chen YC, Hung CH, Cheng JT. Physical Exercise Induces Excess Hsp72 Expression and Delays the Development of Hyperalgesia and Allodynia in Painful Diabetic Neuropathy Rats. Anesthesia & Analgesia 2013; 116(2): 482-90.
17- Yoon H, Thakur V, Isham D, Fayad M, Chattopadhyay M. Moderate Exercise Training Attenuates Inflammatory Mediators in DRG of Type 1 Diabetic Rats. Experimental Neurology 2015; 267: 107-14.
18- Rossi DM, Valenti VE, Navega MT. Exercise Training Attenuates Acute Hyperalgesia in Streptozotocin-Induced Diabetic Female Rats. Clinics (Sao Paulo) 2011; 66(9): 1615-9.
19- Gong YH, Yu XR, Liu HL, Yang N, Zuo PP, Huang YG. Antinociceptive Effects of Combination of Tramadol and Acetaminophen on Painful Diabetic Neuropathy in Streptozotocin-Induced Diabetic Rats. Acta Anaesthesiol Taiwan 2011; 49(1): 16-20.
20- Chae CH, Jung SL, An SH, Jung CK, Nam SN, Kim HT. Treadmill Exercise Suppresses Muscle Cell Apoptosis by Increasing Nerve Growth Factor Levels and Stimulating P-Phosphatidylinositol 3-Kinase Activation in the Soleus of Diabetic Rats. J Physiol Biochem 2011; 67(2): 235-41.
21- Gelderd JB, Chopin SF. The Vertebral Level of Origin of Spinal Nerves in the Rat. Anat Rec 1977; 188(1): 45-7.
22- Keri KC, Samji NS, Blumenthal S. Diabetic Nephropathy: Newer Therapeutic Perspectives. J Community Hosp Intern Med Perspect 2018; 8(4): 200-7.
23- Thakur V, Sadanandan J, Chattopadhyay M. High-Mobility Group Box 1 Protein Signaling in Painful Diabetic Neuropathy. Int J Mol Sci 2020; 21(3): 881.
24- Ismail CAN, Suppian R, Ab Aziz CB, Long I. Expressions of Spinal Microglia Activation, BDNF, and DREAM Proteins Correlated with Formalin-Induced Nociceptive Responses in Painful and Painless Diabetic Neuropathy Rats. Neuropeptides 2020; 79: 102003.
25- Wang D, Couture R, Hong Y. Activated Microglia in the Spinal Cord Underlies Diabetic Neuropathic Pain. Eur J Pharmacol 2014; 728: 59-66.
26- Jin GL, Hong LM, Liu HP, Yue RC, Shen ZC, Yang J, et al. Koumine Modulates Spinal Microglial M1 Polarization and the Inflammatory Response through the Notch-RBP-Jκ Signaling Pathway, Ameliorating Diabetic Neuropathic Pain in Rats. Phytomedicine 2021; 90: 153640.
27- Xiaohua G, Dongdong L, Xiaoting N, Shuoping C, Feixia S, Huajun Y, et al. Severe Vitamin D Deficiency is Associated with Increased Expression of Inflammatory Cytokines in Painful Diabetic Peripheral Neuropathy. Front Nutr 2021; 8: 612068.
28- da Luz Scheffer D, Latini A. Exercise-Induced Immune System Response: Anti-Inflammatory Status on Peripheral and Central Organs. Biochim Biophys Acta Mol Basis Disease 2020; 1866(10): 165823.
29- Sun JS, Yang YJ, Zhang YZ, Huang W, Li ZS, Zhang Y. Minocycline Attenuates Pain by Inhibiting Spinal Microglia Activation in Diabetic Rats. Mol Med Rep 2015; 12(2): 2677-82.
30- Gong X, Chen Y, Fu B, Jiang J, Zhang M. Infant Nerve Injury Induces Delayed Microglial Polarization to The M1 Phenotype, and Exercise Reduces Delayed Neuropathic Pain By Modulating Microglial Activity. Neuroscience 2017; 349: 76-86.
متن کامل: (599 مشاهده)
مقدمه
نوروپاتی دیابتی (Diabetic neuropathy) DN آسیب عصبی بسیار شایع و ناتوانکننده است که بیش از نیمی از بیماران دیابتی را تحت تأثیر قرار میدهد. مشخصه پاتوژنز DN آسیب پیشرونده و از بین رفتن فیبرهای عصبی میلین دار و بدون میلین است که طیف وسیعی از نقایص و علائم حسی را نشان میدهد، این علائم میتوان بهصورت بیحسی، ضعف و درد در اندامهای انتهایی ظاهر شوند (1,2). پژوهشها، اختلال در مسیرهای متابولیکی گلوکز را بهعنوان اصلیترین پاتوژنز DN شناختهاند. محققین نشان دادهاند که هایپر گلیسمی مزمن منجر به ورود و تجمع گلوکز اضافه در نورونها میشود (3). گلوکز مازاد از طریق مسیرهای متابولیکی متناوب موجب دوچرخه معیوب التهاب و استرس اکسیداتیو در سیستم عصبی میشوند (3). تحقیقات زیادی افزایش سطح سایتوکینهای پیش التهابی و شاخصهای استرس اکسیداتیو را در سیستم عصبی بیماران دیابتی نشان دادهاند (1,4,5). مشخصشده است که افزایش سطح این شاخصها در نورونها، منجر به کاهش پرفیوژن اعصاب محیطی شده و درنتیجه آن ایسکمی عصبی رخ میدهد. به دنبال ایسکمی، فعالسازی و تکثیر میکروگلیاها و نفوذ مازاد نوتروفیلها و منوسیتهای گردش خون در اعصاب محیطی افزایش مییابد (5). این عوامل با سازوکارهای متعددی ازجمله تولید گونههای فعال اکسیژن Reactive oxygen species، افزایش سطح سیتوکینها و پروتئازها، موجب آسیب مخروطهای رشدی، انحطاط آکسونی، تخریب غلاف میلین، همچنین اختلال نوزایش عصبی (neurogenesis) و مرگ سلولهای عصبی شده و نوروپاتی دیابتی را ارتقاء میبخشند (5)؛ بنابراین مهار التهاب و استرس اکسیداتیو در بیماران دیابتی برای تخریب پیشرونده بافت عصبی دارای نقش اساسی و مهمی است. اینترلوکین 35 Interleukin 35 (IL-35) یک پروتئین هترودایمری است که از زیر واحدهای ژن EBI3 کدگذاری میشود. این سایتوکین با ظرفیت ضدالتهابی و سرکوبکننده سیستم ایمنی از طریق زیر واحدهای IL-12Rβ2 و IL-27Rα سیگنال میدهد (6,7). تحقیقات کاهش سطوح این پروتئین را در بیماران دیابتی نشان دادهاند (8). شواهد زیادی نشان دادهاند که IL-35 از طریق مبدل سیگنال STAT1 منجر به مهار TNF-α ناشی از فاکتور رونویسی NF-κB شده و از پروسه التهاب جلوگیری میکند (8,9)؛ بنابراین استفاده از راهکارهای غیر دارویی که بتواند این مسیر پیامرسانی را تنظیم مثبت کند ممکن است بهعنوان یک هدف مولکولی برای درمان تخریب عصبی در بیماران مبتلابه نورودژنراتیو ناشی از دیابت مورداستفاده قرار گیرد. نتایج حاصل از مطالعات اخیر نشان داده است فعالیتهای هوازی بهعنوان یک راهبرد غیر دارویی با افزایش سطوح پروتئینهای شوک گرمایی، افزایش سطوح سایتوکینهای ضدالتهابی، کاهش فعالیت میکروگلیاهای نخاع، افزایش بیان نوروترفینها و کاهش سطوح رادیکالهای آزاد در سیستم عصبی توانسته است ظرفیت آنتیاکسیدانی طبیعی سلول را بالابرده و فشار اکسایشی و التهاب عصبی را کاهش دهد درنتیجه از تخریب پیشروندِ نورونهای حسی جلوگیری کند (10,11). با توجه به شواهد زیادی مبنی براثرگذاری تمرین هوازی بر مسیرهای مولکولی و بیوشیمیایی در نوروپاتی دیابتی، اما تاکنون تأثیر ضد دردی تمرین هوازی در رابطه با اینترلوکین 35 و ماهیت ضدالتهابی آن مورد ارزیابی قرار نگرفته است. بنابراین فرض تحقیق بر این است که تمرین هوازی با تنظیم مثبت فعالیت سایتوکین ضدالتهابی IL-35 تأثیر بهسزایی در کاهش سطح التهاب عصبی و درد نوروپاتیک دیابتی ایفا خواهد کرد.
روش بررسی
در پژوهش حاضر راهبرد تحقیق از نوع تجربی بود. تعداد 24 سر موش صحرایی نر نژاد ویستار 8 هفتگی با محدوده وزنی 11±204/3 گرم از مرکز تکثیر حیوانات آزمایشگاهی دانشگاه علوم پزشکی جندیشاپور اهواز تهیه و در گروههای چهارتایی در قفسهای استاندارد پلی کربنات در شرایط دمایی 2±22 درجه سانتیگراد تحت چرخه 12:12 ساعت تاریکی – روشنایی و با دسترسی آزاد به آب و غذای ویژه موش نگهداری شدند. بعد از گذشت یک هفته سازگاری با محیط آزمایشگاه، آشناسازی با نوار گردان و دستکاری، موشها بهطور تصادفی به سه گروه (8 = n) نوروپاتی دیابت دیابتی (تزریق درون صفاقی 50 میلیگرم استرپتوزوسین/کیلوگرم وزن بدن)، نوروپاتی دیابت تمرین (30 دقیقه تمرین هوازی با شدت 15 متر در دقیقه، 5 روز در هفته به مدت 6 هفته) و شاهد تقسیم شدند. در پژوهش حاضر، کلیه اصول اخلاقی کار با حیوانات توسط کمیته اخلاق پژوهشگاه تربیتبدنی و علوم ورزشی با کد (IR.SSRC.REC.1397.017) مورد تائید قرار گرفت.
القاء دیابت: پس از اتمام پروتکل آشناسازی و متعاقب 12 ساعت محرومیت از غذا، القاء دیابت با تزریق درون صفاقی محلول استروپتوزوتوسین Strepotozocin(STZ) (Sigma, St. Louis, MO)؛ حلشده در بافر سیترات 0/05 مولار با 4/5pH: 50 میلیگرم به ازای هر کیلوگرم وزن بدن بهمنظور ایجاد دیابت نوع 1 صورت گرفت (12,13). به موشهای گروه شاهد نیز معادل حجمی بافر سیترات 0/05 مولار با 4/5:pH بهصورت درون صفاقی تزریق شد. 48 ساعت پس از تزریق، با ایجاد یک جراحت کوچک توسط لانست بر روی ورید دم، یک قطره خون بر روی نوار گلوکومتری قرار داده شد و نوار توسط دستگاه گلوکومتر (Glucotrend 2، شرکت روشه آلمان) اندازهگیری و موشهای صحرائی که قند خون آنها بیشتر از mg/dl250 بود، بهعنوان دیابتی در نظر گرفته شدند. برای اطمینان از عدم بازگشت قند خون، در طول دوره برنامه تمرینی هر هفته و نیز پایان دوره، قند خون موشها اندازهگیری شد (14).
آزمونهای رفتاری: پیش از القاء دیابت، بهمنظور سازگاری جهت آزمونهای رفتاری، حیوانات سه روز در معرض آزمایش (دو بار برای هر آزمون) قرار گرفتند. دو هفته پس از القای دیابت، آزمونهای رفتاری درد نوروپاتیک بهعنوان شاخص وقوع شرایط پاتولوژیکی نوروپاتی دیابت و برای تائید و میزان درد نوروپاتیک از تمامی گروهها به عمل آمد (17-15). بهمنظور بررسی اثرات طولانیمدت تمرین هر هفته و تا پایان پروتکل تمرین هوازی آزمونهای رفتاری درد نوروپاتیک اجرا شد، برای اجتناب از عوامل مداخله¬گر نظیر اثرات ضد دردی القاء شده توسط استرس آزمایشهای رفتاری میان ساعت 7 تا 10 صبح انجام شد (17).
آزمون هات پلیت Hot Plate Test: برای اندازهگیری تغییر آستانه درد حرارتی (هایپرآلژزیای حرارتی) از آزمون هاتپلیت استفاده شد. این آزمون بر اساس روش والف و مکدونالد انجام گرفت (18). برای انجام این آزمون، از دستگاه هاتپلیت مدل ام اچ -9500 ساخت شرکت برج صنعت آزما که دارای یک صفحه فلزی به قطر 19 سانتیمتر و محفظهای از جنس پلکسی گلاس (30×25×25 سانتیمتر) استفاده شد. دستگاه مجهز به زمانسنج و ترموستات بود. شدت درجه گرمایی صفحه دستگاه در 2±52 درجه سانتیگراد تنظیم شد. قبل از انجام آزمون، برای آشنایی، موشها را به مدت 2 دقیقه بر روی صفحه دستگاه قرار داده شد؛ سپس دستگاه روشن شد تا دمای صفحه دستگاه به دمای موردنظر ثابت شود. حیوان بر روی صفحه داغ قرار گرفت و همزمان با آن، زمانسنج دستگاه روشن شد. زمانی که حیوان شروع به لیسیدن، بالا بردن و یا لرزیدن پا کرد، بهعنوان نقطه پایانی و شاخص احساس درد تلقی شد و فوراً زمانسنج متوقف و حیوان از دستگاه خارج میشد. مدتزمان تأخیر در پس کشیدن پنجه (Paw withdrawal latency) و یا فاصله زمانی شروع قرار گرفتن حیوان بر روی صفحه داغ تا پیدایش پاسخ به درد توسط حیوان، در سه مرحله و به فاصله زمانی 5 دقیقه در گروههای مختلف برحسب ثانیه اندازهگیری شد؛ و میانگین آنها بهعنوان زمان تأخیر ثبت گردید. زمان عدم واکنش حیوان به صفحه داغ 30 ثانیه (Cut of time) در نظر گرفته شد (19).
آزمون آلودینیای مکانیکی Mechanical allodynia test: بهمنظور اندازهگیری تعیین آستانه درد، حیوان روی یک شبکۀ سیمی و داخل یک محفظۀ پلکسی گلاس به ابعاد 20×20 و ارتفاع 30 سانتیمتر قرار گرفت. جهت عادت کردن حیوانات به محیط جدید 30 دقیقه قبل از آزمایش، درون محفظه شفاف و روی صفحۀ مشبک قرار گرفتند. در ادامه بهمنظور سنجش آلودینیای مکانیکی، از تارهای مختلف Von Frey در محدوده 2 تا 60 گرم (60، 26، 15، 10، 8، 6، 4، 2) (ساخت شرکت استولتین کشورآمریکا) جهت سنجش حساسیت پوست به تحریکات تماسی استفاده شد. هر آزمایش با تار دارای کمترین وزن شروع میشد و در صورت عدم ایجاد پاسخ، به ترتیب از تارهای با وزن بالاتر استفاده میگردید. همچنین چنانچه دو بار متوالی پاسخ (بلند کردن پا توسط حیوان) مشاهده میگردید. همان وزنه بهعنوان آستانه پس کشیدن پنجه Paw withdrawal threshold (PWT) ثبت میشد و آزمون خاتمه مییافت. در مقابل، اگر حیوان به هیچیک از تارها ازجمله تار شماره 60 پاسخ نمیداد، عدد 60 بهعنوان آستانۀ پاسخ در نظر گرفته میشد. هر آزمایش سه بار و بهتناوب حداقل سه دقیقه تکرار شد و میانگین آنها بهعنوان آستانۀ پس کشیدن پنجه منظور گردید (20).
پروتکل تمرین هوازی: پس از اطمینان از حصول نوروپاتی دیابت در موشهای صحرایینر، پروتکل تمرین هوازی به مدت شش هفته اجرا شد. در پژوهش حاضر پروتکل تمرین هوازی بر اساس مطالعه چانگ هوان و همکاران (2011) انجام گرفت؛ ابتدا بهمنظور خوگیری به شرایط آزمایشگاه، نوار گردان و دستکاری حیوانات 5 روز در هفته به مدت 10 -15 دقیقه و با سرعت 10 متر بر دقیقه بروی نوار گردان راه رفتند. سپس گروه ورزشی نوروپاتی دیابت تمرین در معرض تمرین نوار گردان، 5 جلسه در هفته و به مدت 6 هفته قرار گرفت (21). سرعت و مدت تمرین نوار گردان هر هفته بهتدریج افزایش یافت و از 10 متر در دقیقه به مدت 10 دقیقه در هفته اول، 10 متر در دقیقه برای 20 دقیقه در هفته دوم، 14 تا 15 متر در دقیقه برای 20 دقیقه در هفته سوم، 14 تا 15 متر در دقیقه برای 30 دقیقه در هفته چهارم و 17 تا 18 متر در دقیقه برای 30 دقیقه در هفته پنجم و ششم افزایش یافت. جهت رسیدن سازگاریهای بهدستآمده به حالت یکنواخت، تمامی متغیرهای تمرینی در هفته پایانی (هفته ششم) ثابت نگهداشته شد. تمام جلسات تمرینی در پایان سیکل خواب حیوانات و بین ساعت 18-16 عصر برگزار شد.
استخراج نمونه و روش اندازهگیری: در پایان شش هفته برنامه تمرینی، 48 ساعت پس از آخرین جلسه تمرین، موش¬ها بهوسیلهی تزریق درون صفاقی ترکیبی از کتامین (90میلیگرم به ازای هر کیلوگرم وزن) و زایلازین (10 میلیگرم به ازای هر کیلوگرم وزن) بیهوش شدند. برای بررسی متغیرهای بیوشیمیایی، تحت شرایط سترون و مطابق روش گلدرد و چوپین سال 1977، (22) ابتدا قطعه نخاعی از سطح L4 تا L6 (سگمنت¬های نخاعی مربوطه در موشهای نر نژاد ویستار در ناحیه مهرههای T13-L1 ستون فقرات قرار دارد) مشخص گردید، سپس با برش در پایینترین بخش ممکن از ستون فقرات جدا شد. ستون فقرات با استفاده از کانال مرکزی بهعنوان شاخص، به بخش قدامی و خلفی تفکیک شد و بخش خلفی نخاع در ناحیه مربوطه را بهعنوان نمونه، در نیتروژن مایع منجمد و نمونهها تا زمان انجام آزمایشهای مولکولی در فریزر 80- درجه سانتیگراد نگهداری شدند.
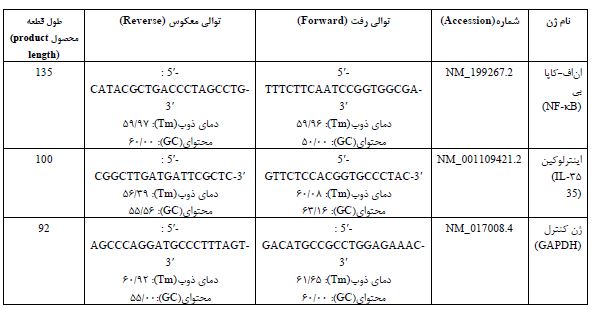
Real Time-PCR: بهمنظور استخراج RNA، 50 میلیگرم از بافت نخاع با اضافه کردن 300 میکرو لیتر ترایزول (کیاژن، آمریکا) هموژن گردید. مراحل مختلف طبق دستورالعمل کیت استخراج RNA تا مرحله نهایی و تهیه RNA خالص انجام شد. محلول RNA استخراجشده با آنزیم DNase I از هرگونه آلودگی به DNA و آنزیمهای تخریبکننده RNA پاکسازی شد. نسبت جذبی 260 به 280 نانومتر به شیوه طیفسنجی نوری برای تمامی نمونههای استخراج شده بین 1/8 تا 2 بود. بهمنظور سنتز cDNA، 5 میکروگرم از RNA استخراج شده با استفاده از دستورالعمل کیت RevertAsid First Strand cDNA Synthesis (ترموساینتیفیک آمریکا) و با استفاده از پرایمرهای سیناکلون به cDNA تبدیل گردید. از تکنیک RT-qPCR جهت -بررسی بیان ژنهای NF-κB و IL-35 بهصورت کمی استفاده شد، هر واکنش PCR با استفاده از دستگاه (PCR master mix Applied Biosystems) و SYBR Green در دستگاهABI Step One (Applied Biosystems, Sequence Detection Systems. Foster City, CA) طبق برنامه شرکت سازنده انجام گرفت. چهل چرخه دمایی برای هر فرایند Real-Time PCR در نظر گرفته شد و دماهای هر چرخه شامل 94 درجه سانتیگراد برای 20 ثانیه، 60-58 درجه سانتی¬گراد برای 30 ثانیه و 72 درجه سانتیگراد برای 30 ثانیه تنظیم شدند. ضمن اینکه از GAPDH بهعنوان ژن مرجع استفاده گردید. نسبت بیان ژنهای مورد بررسی در این مطالعه، با روش مقایسهای چرخه آستانه Thereshold Cycle(CT) مورد ارزیابی قرار گرفتند. با استفاده از قرار دادن دادهها در فرمول R=2^(- (∆∆CT)) میزان بیان ژن هدف با ژن مرجع نرمالیز شده و بیان ژنهای گروه سالم بهعنوان کالیبراتور در نظر گرفته شد. مشخصات پرایمرهای سنتز شده در جدول 1 ذکرشده است.
تجزیهو تحلیل آماری
جهت تعیین نرمال بودن دادهها از آزمون کلوموگروف- اسمیرنوف Kolmogorov–Smirnov test استفاده شد. برای بررسی معنیدار بودن اختلاف بین گروهها از تحلیل واریانس یکطرفه و در صورت معنیداری، جهت تعیین تفاوت بین میانگینهای دوگروهی از آزمون تعقیبی توکی استفاده شد. تجزیه و تحلیل دادهها با استفاده از نرمافزارversion 16 SPSS در سطح معنیداری 0/5 (0/05>p) انجام شد.
ملاحظات اخلاقی
پروپوزال این تحقیق توسط دانشگاه لرستان با کد اخلاق (IR.SSRC.REC.1397.017) تایید شده است
نتایج
نتایج نشان داد که وزن اولیه گروهها اختلاف معناداری با یکدیگر نداشت (0/328=P)، اما در هفتههای پایانی پژوهش، میانگین تغییرات وزن موشهای گروههای نوروپاتی دیابت نسبت به گروه شاهد بهصورت معناداری کمتر بود (0/001=P). همچنین، میانگین تغییرات وزن گروه نوروپاتی دیابت تمرین نسبت به گروه نوروپاتی دیابت، در هفته هشتم افزایش معنادار داشتند (0/063=P) (جدول 2). پس از القاء دیابت، سطوح گلوکز خون بهصورت معناداری در گروههای نوروپاتی دیابت افزایش یافت (0/001=P) و این اختلاف تا پایان دوره پژوهش در مقایسه با گروه شاهد همچنان معنیدار بود (0/001=P)، همچنین، در پایان برنامه تمرینی، غلظت گلوکز خون گروه نوروپاتی دیابت تمرین نسبت به گروه نوروپاتی دیابت بهصورت معناداری پایینتر بود (0/001=P) ولی بااینحال همچنان اختلاف معنیداری با گروه شاهد وجود داشت (0/012=P) (جدول 2). میانگین مدتزمان تأخیر در پس کشیدن پنجه (Paw withdrawal latency) در آزمون هات پلیت دو هفته پس از القاء دیابت در گروههای نوروپاتی دیابت نسبت به گروه شاهد بهطور معنیداری کمتر بود (0/001=P). همچنین در هفتههای پایانی اجرای پروتکل تمرین هوازی، میانگین مدتزمان تأخیر در پس کشیدن پنجه در آزمون هایپرآلژزیای حرارتی در گروه نوروپاتی دیابت تمرین نسبت به گروه نوروپاتی دیابت بهطور معنیداری بیشتر بود (0/001=P) (نمودار 1).
دو هفته بعد از القاء دیابت، میانگین تغییرات آستانه پس کشیدن پنجه در آزمون آلودینیای مکانیکی در گروه¬های نوروپاتی دیابت نسبت به گروه شاهد بهطور معنی¬داری کمتر بود (0/005=P). از طرفی در هفتههای هفتم و هشتم اجرای پروتکل، میانگین تغییرات آزمون آلودینیای مکانیکی در گروه نوروپاتی دیابت تمرین افزایش معنیداری نسبت به گروه نوروپاتی دیابت داشتند (026/0=P) (نمودار2). با توجه به میانگین گروهها، مشخص شد که القاء دیابت موجب کاهش معنادار در میزان بیان ژن IL-35 و افزایش معناداری در ژن NF-kB در بخش خلفی نخاع موشهای نوروپاتی دیابت در مقایسه با گروه شاهد شد (0/001=P) (P=0/021). میزان بیان ژن IL-35 در گروه نوروپاتی دیابت تمرین بهطور معناداری نسبت به گروه نوروپاتی دیابت بالاتر بود (0/007=P). همچنین میزان بیان ژن NF-kB در گروه نوروپاتی دیابتی تمرین بهطور معناداری نسبت به گروه نوروپاتی دیابت پائینتر بود (0/034=P). از طرفی میزان بیان ژنهای فوق بین گروهای نوروپاتی دیابت تمرین و گروه شاهد تفاوت معنیدار نشان داده نشد (0/2=P) (نمودار 3).
جدول 1: مشخصات توالی پرایمر های ژنهای مورداستفاده در پژوهش
جدول 2: نتایج آزمون تحلیل واریانس دو طرفه وزن بدن و سطح گلوکز خون در موشهای گروههای مختلف

کلیه مقادیر جدول بهصورت انحراف استاندارد ± میانگین میباشند. *اختلاف معنیدار با گروه شاهد (0/05>p). †اختلاف معنیدار با گروه نوروپاتی دیابتی (0/05>p).
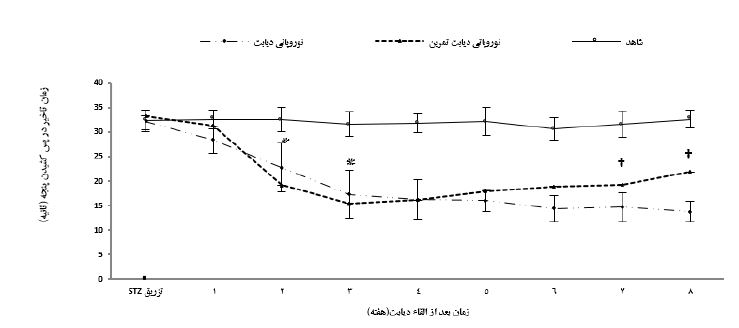
نمودار 1: تغییرات زمان تأخیر در پس کشیدن پنجه در آزمون هایپرآلژزیای حرارتی گروههای مختلف
* اختلاف معنیدار با گروه شاهد P<0.05)). † اختلاف معنیدار با گروه نوروپاتی دیابتی P<0.05)).
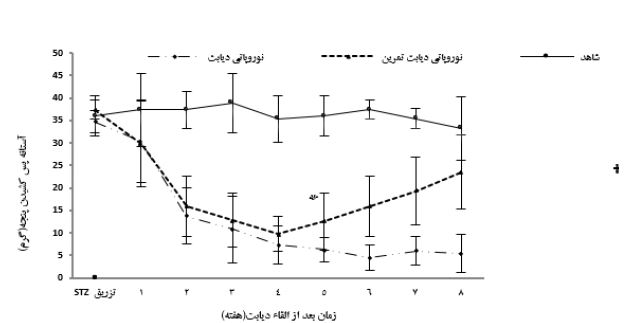
نمودار 2: تغییرات آستانه پس کشیدن پنجه در آزمون آلودینیای مکانیکی گروههای مختلف
* اختلاف معنیدار با گروه شاهد P<0.05)). † اختلاف معنیدار با گروه نوروپاتی دیابتی P<0.05)).
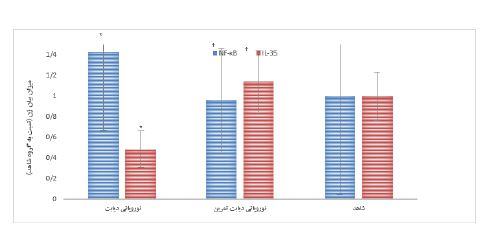
نمودار 3: میزان بیان ژن NF-κB و IL-35 در بخش خلفی نخاع گروههای مختلف.
* اختلاف معنیدار با گروه شاهد P<0.05)). † اختلاف معنیدار با صگروه نوروپاتی دیابتی P<0.05)).
بحث
در پژوهش حاضر اثر تمرین هوازی بر میزان بیان ژن IL-35 و NF-kB در بافت نخاع موشهای مبتلابه درد نوروپاتی دیابتی و همچنین آستانه پاسخ به آزمونهای رفتاری درد نوروپاتی، آزمونهات پلیت (حساسیت سیستم عصبی به محرکهای دردناک) و آزمون آلودینیای مکانیکی (حساسیت سیستم عصبی به محرکهای بدون درد) مورد بررسی قرار گرفت. یافتههای پژوهش حاضر نشان داد که پس از القاء و اثبات درد نوروپاتی دیابت ناشی از تزریق استرپتوزوسین، تمرین هوازی، میزان بیان ژن IL-35 را در بخش خلفی نخاع بهطور معنیداری افزایش و میزان بیان ژن NF-kB را کاهش و آستانه پاسخ به آزمونهای رفتاری درد نوروپاتی را افزایش داده است. تحقیقات زیادی نشان دادهاند که اتو اکسیداسیون گلوکز به دنبال هیپرگلیسمی باعث تولید بیرویه رادیکالهای آزاد و سطوح عوامل التهابی در سلولها ازجمله نورونها میشود، گمان میرود که این تغییرات بیوشیمیایی در سلولهای گلیال و نورونهای محیطی باعث اختلال در حساسیت طبیعی نوسیسپتورها به محرکهای درد زا و غیر درد زا میشود (23). بهطورکلی پذیرفتهشده است که تولید بیشازحد واسطههای التهابی درنتیجه دیابت، نقش مهمی در شروع و حفظ درد نوروپاتی دیابتی دارد (24). در شرایط فیزیولوژی طبیعی، میکروگلیاها واسطههای ضدالتهابی را آزاد و بقایای سلولهای سمی را فاگوسیتوز کرده تا هموستاز را در سیستم عصبی حفظ کنند. مشخص شده است که میکروگلیاها توسط عوامل متعددی از جمله هایپرگلیسمی فعال میشوند، فعالسازی طولانیمدت میکروگلیاها باعث تولید بیشازحد سیتوکین های التهابی شده که این عامل منجر به التهاب عصبی میشود (25). پژوهشها نشان دادند که تحت شرایط استرس ناشی از هایپرگلیسمی، فنوتیپ سلولهای میکروگلیال به M1 تغییر کرده و میزان بیان ژنهای پیش التهابی IL-6، IL-1β و TNF-α افزایش مییابند، این عامل نقش مهمی در شروع و تداوم درد نوروپاتیک دیابتی دارد (26). تحقیقات اخیر گزارش دادهاند که عواملی میتوانند پلاریزاسیون میکروگلیالها از فنوتیپ M1 پیش التهابی به فنوتیپ ضدالتهابی M2 را تغییر دهند، شناسایی و بهکارگیری این عوامل اثرگذار در این تغییر نشاندهنده یک راهبرد درمانی جدید برای درد نوروپاتی دیابتی است. محققین افزایش سطوح عوامل التهابی CD68، iNOS، IL-6، IL-1β و TNF-α را بهعنوان نشانگرهای فعالیت فنوتیپ M1 میکروگلیالها عنوان کردهاند (9,27). نتایج حاصل از دادههای پژوهش حاضر نشان داد که میزان بیان ژن فاکتور رونویسی NF-kB، القاء کننده عوامل التهابی در گروه نوروپاتی دیابتی افزایش بیانشده بود. افزایش بیان این عامل رونویسی در نخاع، نشان بر فعالیت میکروگلیال ها فنوتیپ M1 دارد. آزمایشهای حیوانی و مطالعات In Vitro نشان دادهاند که IL-35 برونزا از طریق مهار مسیر پیامرسانی JNK و تنظیم مثبت مسیر سیگنالینگ JAK2/STAT6 نقش مهمی در پلاریزاسیون ماکروفاژها از فنوتیپ M1 التهابی به فنوتیپ ضدالتهابی M2 ایفا میکند (8,9). با توجه به اینکه افزایش نشانگرهای فعالیت میکروگلیال های فنوتیپ M2 شاخصهای ضدالتهابی همانند ARG-1، IL-10 و IL-35 هستند (9). احتمالاً کاهش میزان بیان ژن IL-35 در بافت نخاع موشهای صحرایی گروه نوروپاتی دیابتی در پژوهش حاضر، کاهش فعالیت فنوتیپ M2 را در این گروه نشان دهد. تحقیقات نشان دادهاند که سلولهای سیستم ایمنی اثر بیولوژیکی خود را از طریق تنظیم بیان سایتوکین های ضدالتهابی مانند IL-10، IL-4 و IL-35 انجام میدهند (28)؛ بنابراین هدف قرار دادن سایتوکین ضدالتهابی IL-35 و سنتز آن در نورونهای حسی، ممکن است، بهعنوان یک رویکرد مهم درمانی برای درد نوروپاتیک دیابت استفاده شود. مطالعات اثرات محافظتی تمرین هوازی را در طی شرایط فیزیولوژیکی و پاتولوژیکی ازجمله تنظیم سیستم ایمنی، افزایش سطوح شاخصهای ضدالتهابی و افزایش ظرفیت آنتیاکسیدانی بهخوبی به اثبات رساندهاند (29). ولی هیچگونه مطالعهای، نقش فعالیت هوازی را بر سایتوکین ضدالتهابی IL-35 در سیستم عصبی محیطی و رابطه آن با آزمونهای رفتاری درد نورپاتیک انجام نگرفته است. مطالعات نشان دادهاند که فعالیتهای ورزشی قطبش میکروگلیال های نخاع را به فنوتیپ M2 تغییر داده و نوروپاتی دیابتی را بهبود بخشیده است (30)؛ بنابراین این احتمال وجود دارد که فعالیت هوازی، بهواسطه اثرات آنتیاکسیدانی و ضدالتهابی، توانسته است تولید پروتئینهای آنتیاکسیدان و عوامل ضدالتهابی مانند IL-35 را تنظیم مثبت کرده و از این طریق با تأثیر و دخالت بر مهار مسیرهای التهابی در سیستم عصبی، باعث کاهش عوارض نوروپاتی دیابتی در موشهای صحرایی مورد آزمون شده باشد. بااینحال، عدم اندازهگیری پروتئینهای دفاعی آنتیاکسیدانهای آنزیمی و غیر آنزیمی که در نتیجه نوروپاتی دیابتی تنظیم مثبت میشوند، در پژوهش حاضر بهعنوان یک محدودیت از بحث دقیق در این زمینه جلوگیری میکند. با این وجود پیشنهاد میشود در پژوهشهای آتی، همراه با اندازه¬گیری سایتوکین ضدالتهابی IL-35 سایر عوامل ضدالتهابی مانند IL-10 و IL-4 بهعنوان شاخصهای فعالیت میکروگلیاهای فنوتیپ M2 و مارکرهای فنوتیپ M1 شامل CD68، iNOS، IL-6، IL-1β و TNF-α برای دستیابی به شرایط قطعیتر مورد بررسی قرار دهند.
نتیجه گیری
پژوهش حاضر نشان داد که القاء دیابت باعث کاهش میزان بیان ژن IL-35 و افزایش بیان NF-kB در بخش حسی نخاع شده و این عامل منجر به افزایش حساسیت گیرندههای درد میشود. بااینحال شش هفته تمرین هوازی توانسته است این میزان بیان سایتوکین ضدالتهابی IL-35 را تنظیم مثبت و بیان فاکتور رونویسی NF-kB را تنظیم منفی کند و حساسیت سیستم عصبی به محرکهای دردناک را کاهش دهد. لذا بهنظر میرسد که تمرین هوازی از طریق افزایش سطوح سایتوکینهای ضدالتهابی منجر به کاهش التهاب عصبی شده و از این طریق در کاهش عوارض نوروپاتی دیابتی اثرگذار بوده است.
سپاسگزاری
این مقاله مستخرج از پایاننامه دورۀ دکتری رشته فیزیولوژی ورزشی میباشد؛ از کلیه کارشناسان محترم آزمایشگاه و اساتید گروه فیزیولوژی ورزشی دانشگاه لرستان تشکر و قدردانی میشود. ضمناً کلیه هزینههای این طرح شخصی تأمینشده است.
حامی مالی: ندارد.
تعارض منافع: وجود ندارد.
نوروپاتی دیابتی (Diabetic neuropathy) DN آسیب عصبی بسیار شایع و ناتوانکننده است که بیش از نیمی از بیماران دیابتی را تحت تأثیر قرار میدهد. مشخصه پاتوژنز DN آسیب پیشرونده و از بین رفتن فیبرهای عصبی میلین دار و بدون میلین است که طیف وسیعی از نقایص و علائم حسی را نشان میدهد، این علائم میتوان بهصورت بیحسی، ضعف و درد در اندامهای انتهایی ظاهر شوند (1,2). پژوهشها، اختلال در مسیرهای متابولیکی گلوکز را بهعنوان اصلیترین پاتوژنز DN شناختهاند. محققین نشان دادهاند که هایپر گلیسمی مزمن منجر به ورود و تجمع گلوکز اضافه در نورونها میشود (3). گلوکز مازاد از طریق مسیرهای متابولیکی متناوب موجب دوچرخه معیوب التهاب و استرس اکسیداتیو در سیستم عصبی میشوند (3). تحقیقات زیادی افزایش سطح سایتوکینهای پیش التهابی و شاخصهای استرس اکسیداتیو را در سیستم عصبی بیماران دیابتی نشان دادهاند (1,4,5). مشخصشده است که افزایش سطح این شاخصها در نورونها، منجر به کاهش پرفیوژن اعصاب محیطی شده و درنتیجه آن ایسکمی عصبی رخ میدهد. به دنبال ایسکمی، فعالسازی و تکثیر میکروگلیاها و نفوذ مازاد نوتروفیلها و منوسیتهای گردش خون در اعصاب محیطی افزایش مییابد (5). این عوامل با سازوکارهای متعددی ازجمله تولید گونههای فعال اکسیژن Reactive oxygen species، افزایش سطح سیتوکینها و پروتئازها، موجب آسیب مخروطهای رشدی، انحطاط آکسونی، تخریب غلاف میلین، همچنین اختلال نوزایش عصبی (neurogenesis) و مرگ سلولهای عصبی شده و نوروپاتی دیابتی را ارتقاء میبخشند (5)؛ بنابراین مهار التهاب و استرس اکسیداتیو در بیماران دیابتی برای تخریب پیشرونده بافت عصبی دارای نقش اساسی و مهمی است. اینترلوکین 35 Interleukin 35 (IL-35) یک پروتئین هترودایمری است که از زیر واحدهای ژن EBI3 کدگذاری میشود. این سایتوکین با ظرفیت ضدالتهابی و سرکوبکننده سیستم ایمنی از طریق زیر واحدهای IL-12Rβ2 و IL-27Rα سیگنال میدهد (6,7). تحقیقات کاهش سطوح این پروتئین را در بیماران دیابتی نشان دادهاند (8). شواهد زیادی نشان دادهاند که IL-35 از طریق مبدل سیگنال STAT1 منجر به مهار TNF-α ناشی از فاکتور رونویسی NF-κB شده و از پروسه التهاب جلوگیری میکند (8,9)؛ بنابراین استفاده از راهکارهای غیر دارویی که بتواند این مسیر پیامرسانی را تنظیم مثبت کند ممکن است بهعنوان یک هدف مولکولی برای درمان تخریب عصبی در بیماران مبتلابه نورودژنراتیو ناشی از دیابت مورداستفاده قرار گیرد. نتایج حاصل از مطالعات اخیر نشان داده است فعالیتهای هوازی بهعنوان یک راهبرد غیر دارویی با افزایش سطوح پروتئینهای شوک گرمایی، افزایش سطوح سایتوکینهای ضدالتهابی، کاهش فعالیت میکروگلیاهای نخاع، افزایش بیان نوروترفینها و کاهش سطوح رادیکالهای آزاد در سیستم عصبی توانسته است ظرفیت آنتیاکسیدانی طبیعی سلول را بالابرده و فشار اکسایشی و التهاب عصبی را کاهش دهد درنتیجه از تخریب پیشروندِ نورونهای حسی جلوگیری کند (10,11). با توجه به شواهد زیادی مبنی براثرگذاری تمرین هوازی بر مسیرهای مولکولی و بیوشیمیایی در نوروپاتی دیابتی، اما تاکنون تأثیر ضد دردی تمرین هوازی در رابطه با اینترلوکین 35 و ماهیت ضدالتهابی آن مورد ارزیابی قرار نگرفته است. بنابراین فرض تحقیق بر این است که تمرین هوازی با تنظیم مثبت فعالیت سایتوکین ضدالتهابی IL-35 تأثیر بهسزایی در کاهش سطح التهاب عصبی و درد نوروپاتیک دیابتی ایفا خواهد کرد.
روش بررسی
در پژوهش حاضر راهبرد تحقیق از نوع تجربی بود. تعداد 24 سر موش صحرایی نر نژاد ویستار 8 هفتگی با محدوده وزنی 11±204/3 گرم از مرکز تکثیر حیوانات آزمایشگاهی دانشگاه علوم پزشکی جندیشاپور اهواز تهیه و در گروههای چهارتایی در قفسهای استاندارد پلی کربنات در شرایط دمایی 2±22 درجه سانتیگراد تحت چرخه 12:12 ساعت تاریکی – روشنایی و با دسترسی آزاد به آب و غذای ویژه موش نگهداری شدند. بعد از گذشت یک هفته سازگاری با محیط آزمایشگاه، آشناسازی با نوار گردان و دستکاری، موشها بهطور تصادفی به سه گروه (8 = n) نوروپاتی دیابت دیابتی (تزریق درون صفاقی 50 میلیگرم استرپتوزوسین/کیلوگرم وزن بدن)، نوروپاتی دیابت تمرین (30 دقیقه تمرین هوازی با شدت 15 متر در دقیقه، 5 روز در هفته به مدت 6 هفته) و شاهد تقسیم شدند. در پژوهش حاضر، کلیه اصول اخلاقی کار با حیوانات توسط کمیته اخلاق پژوهشگاه تربیتبدنی و علوم ورزشی با کد (IR.SSRC.REC.1397.017) مورد تائید قرار گرفت.
القاء دیابت: پس از اتمام پروتکل آشناسازی و متعاقب 12 ساعت محرومیت از غذا، القاء دیابت با تزریق درون صفاقی محلول استروپتوزوتوسین Strepotozocin(STZ) (Sigma, St. Louis, MO)؛ حلشده در بافر سیترات 0/05 مولار با 4/5pH: 50 میلیگرم به ازای هر کیلوگرم وزن بدن بهمنظور ایجاد دیابت نوع 1 صورت گرفت (12,13). به موشهای گروه شاهد نیز معادل حجمی بافر سیترات 0/05 مولار با 4/5:pH بهصورت درون صفاقی تزریق شد. 48 ساعت پس از تزریق، با ایجاد یک جراحت کوچک توسط لانست بر روی ورید دم، یک قطره خون بر روی نوار گلوکومتری قرار داده شد و نوار توسط دستگاه گلوکومتر (Glucotrend 2، شرکت روشه آلمان) اندازهگیری و موشهای صحرائی که قند خون آنها بیشتر از mg/dl250 بود، بهعنوان دیابتی در نظر گرفته شدند. برای اطمینان از عدم بازگشت قند خون، در طول دوره برنامه تمرینی هر هفته و نیز پایان دوره، قند خون موشها اندازهگیری شد (14).
آزمونهای رفتاری: پیش از القاء دیابت، بهمنظور سازگاری جهت آزمونهای رفتاری، حیوانات سه روز در معرض آزمایش (دو بار برای هر آزمون) قرار گرفتند. دو هفته پس از القای دیابت، آزمونهای رفتاری درد نوروپاتیک بهعنوان شاخص وقوع شرایط پاتولوژیکی نوروپاتی دیابت و برای تائید و میزان درد نوروپاتیک از تمامی گروهها به عمل آمد (17-15). بهمنظور بررسی اثرات طولانیمدت تمرین هر هفته و تا پایان پروتکل تمرین هوازی آزمونهای رفتاری درد نوروپاتیک اجرا شد، برای اجتناب از عوامل مداخله¬گر نظیر اثرات ضد دردی القاء شده توسط استرس آزمایشهای رفتاری میان ساعت 7 تا 10 صبح انجام شد (17).
آزمون هات پلیت Hot Plate Test: برای اندازهگیری تغییر آستانه درد حرارتی (هایپرآلژزیای حرارتی) از آزمون هاتپلیت استفاده شد. این آزمون بر اساس روش والف و مکدونالد انجام گرفت (18). برای انجام این آزمون، از دستگاه هاتپلیت مدل ام اچ -9500 ساخت شرکت برج صنعت آزما که دارای یک صفحه فلزی به قطر 19 سانتیمتر و محفظهای از جنس پلکسی گلاس (30×25×25 سانتیمتر) استفاده شد. دستگاه مجهز به زمانسنج و ترموستات بود. شدت درجه گرمایی صفحه دستگاه در 2±52 درجه سانتیگراد تنظیم شد. قبل از انجام آزمون، برای آشنایی، موشها را به مدت 2 دقیقه بر روی صفحه دستگاه قرار داده شد؛ سپس دستگاه روشن شد تا دمای صفحه دستگاه به دمای موردنظر ثابت شود. حیوان بر روی صفحه داغ قرار گرفت و همزمان با آن، زمانسنج دستگاه روشن شد. زمانی که حیوان شروع به لیسیدن، بالا بردن و یا لرزیدن پا کرد، بهعنوان نقطه پایانی و شاخص احساس درد تلقی شد و فوراً زمانسنج متوقف و حیوان از دستگاه خارج میشد. مدتزمان تأخیر در پس کشیدن پنجه (Paw withdrawal latency) و یا فاصله زمانی شروع قرار گرفتن حیوان بر روی صفحه داغ تا پیدایش پاسخ به درد توسط حیوان، در سه مرحله و به فاصله زمانی 5 دقیقه در گروههای مختلف برحسب ثانیه اندازهگیری شد؛ و میانگین آنها بهعنوان زمان تأخیر ثبت گردید. زمان عدم واکنش حیوان به صفحه داغ 30 ثانیه (Cut of time) در نظر گرفته شد (19).
آزمون آلودینیای مکانیکی Mechanical allodynia test: بهمنظور اندازهگیری تعیین آستانه درد، حیوان روی یک شبکۀ سیمی و داخل یک محفظۀ پلکسی گلاس به ابعاد 20×20 و ارتفاع 30 سانتیمتر قرار گرفت. جهت عادت کردن حیوانات به محیط جدید 30 دقیقه قبل از آزمایش، درون محفظه شفاف و روی صفحۀ مشبک قرار گرفتند. در ادامه بهمنظور سنجش آلودینیای مکانیکی، از تارهای مختلف Von Frey در محدوده 2 تا 60 گرم (60، 26، 15، 10، 8، 6، 4، 2) (ساخت شرکت استولتین کشورآمریکا) جهت سنجش حساسیت پوست به تحریکات تماسی استفاده شد. هر آزمایش با تار دارای کمترین وزن شروع میشد و در صورت عدم ایجاد پاسخ، به ترتیب از تارهای با وزن بالاتر استفاده میگردید. همچنین چنانچه دو بار متوالی پاسخ (بلند کردن پا توسط حیوان) مشاهده میگردید. همان وزنه بهعنوان آستانه پس کشیدن پنجه Paw withdrawal threshold (PWT) ثبت میشد و آزمون خاتمه مییافت. در مقابل، اگر حیوان به هیچیک از تارها ازجمله تار شماره 60 پاسخ نمیداد، عدد 60 بهعنوان آستانۀ پاسخ در نظر گرفته میشد. هر آزمایش سه بار و بهتناوب حداقل سه دقیقه تکرار شد و میانگین آنها بهعنوان آستانۀ پس کشیدن پنجه منظور گردید (20).
پروتکل تمرین هوازی: پس از اطمینان از حصول نوروپاتی دیابت در موشهای صحرایینر، پروتکل تمرین هوازی به مدت شش هفته اجرا شد. در پژوهش حاضر پروتکل تمرین هوازی بر اساس مطالعه چانگ هوان و همکاران (2011) انجام گرفت؛ ابتدا بهمنظور خوگیری به شرایط آزمایشگاه، نوار گردان و دستکاری حیوانات 5 روز در هفته به مدت 10 -15 دقیقه و با سرعت 10 متر بر دقیقه بروی نوار گردان راه رفتند. سپس گروه ورزشی نوروپاتی دیابت تمرین در معرض تمرین نوار گردان، 5 جلسه در هفته و به مدت 6 هفته قرار گرفت (21). سرعت و مدت تمرین نوار گردان هر هفته بهتدریج افزایش یافت و از 10 متر در دقیقه به مدت 10 دقیقه در هفته اول، 10 متر در دقیقه برای 20 دقیقه در هفته دوم، 14 تا 15 متر در دقیقه برای 20 دقیقه در هفته سوم، 14 تا 15 متر در دقیقه برای 30 دقیقه در هفته چهارم و 17 تا 18 متر در دقیقه برای 30 دقیقه در هفته پنجم و ششم افزایش یافت. جهت رسیدن سازگاریهای بهدستآمده به حالت یکنواخت، تمامی متغیرهای تمرینی در هفته پایانی (هفته ششم) ثابت نگهداشته شد. تمام جلسات تمرینی در پایان سیکل خواب حیوانات و بین ساعت 18-16 عصر برگزار شد.
استخراج نمونه و روش اندازهگیری: در پایان شش هفته برنامه تمرینی، 48 ساعت پس از آخرین جلسه تمرین، موش¬ها بهوسیلهی تزریق درون صفاقی ترکیبی از کتامین (90میلیگرم به ازای هر کیلوگرم وزن) و زایلازین (10 میلیگرم به ازای هر کیلوگرم وزن) بیهوش شدند. برای بررسی متغیرهای بیوشیمیایی، تحت شرایط سترون و مطابق روش گلدرد و چوپین سال 1977، (22) ابتدا قطعه نخاعی از سطح L4 تا L6 (سگمنت¬های نخاعی مربوطه در موشهای نر نژاد ویستار در ناحیه مهرههای T13-L1 ستون فقرات قرار دارد) مشخص گردید، سپس با برش در پایینترین بخش ممکن از ستون فقرات جدا شد. ستون فقرات با استفاده از کانال مرکزی بهعنوان شاخص، به بخش قدامی و خلفی تفکیک شد و بخش خلفی نخاع در ناحیه مربوطه را بهعنوان نمونه، در نیتروژن مایع منجمد و نمونهها تا زمان انجام آزمایشهای مولکولی در فریزر 80- درجه سانتیگراد نگهداری شدند.
Real Time-PCR: بهمنظور استخراج RNA، 50 میلیگرم از بافت نخاع با اضافه کردن 300 میکرو لیتر ترایزول (کیاژن، آمریکا) هموژن گردید. مراحل مختلف طبق دستورالعمل کیت استخراج RNA تا مرحله نهایی و تهیه RNA خالص انجام شد. محلول RNA استخراجشده با آنزیم DNase I از هرگونه آلودگی به DNA و آنزیمهای تخریبکننده RNA پاکسازی شد. نسبت جذبی 260 به 280 نانومتر به شیوه طیفسنجی نوری برای تمامی نمونههای استخراج شده بین 1/8 تا 2 بود. بهمنظور سنتز cDNA، 5 میکروگرم از RNA استخراج شده با استفاده از دستورالعمل کیت RevertAsid First Strand cDNA Synthesis (ترموساینتیفیک آمریکا) و با استفاده از پرایمرهای سیناکلون به cDNA تبدیل گردید. از تکنیک RT-qPCR جهت -بررسی بیان ژنهای NF-κB و IL-35 بهصورت کمی استفاده شد، هر واکنش PCR با استفاده از دستگاه (PCR master mix Applied Biosystems) و SYBR Green در دستگاهABI Step One (Applied Biosystems, Sequence Detection Systems. Foster City, CA) طبق برنامه شرکت سازنده انجام گرفت. چهل چرخه دمایی برای هر فرایند Real-Time PCR در نظر گرفته شد و دماهای هر چرخه شامل 94 درجه سانتیگراد برای 20 ثانیه، 60-58 درجه سانتی¬گراد برای 30 ثانیه و 72 درجه سانتیگراد برای 30 ثانیه تنظیم شدند. ضمن اینکه از GAPDH بهعنوان ژن مرجع استفاده گردید. نسبت بیان ژنهای مورد بررسی در این مطالعه، با روش مقایسهای چرخه آستانه Thereshold Cycle(CT) مورد ارزیابی قرار گرفتند. با استفاده از قرار دادن دادهها در فرمول R=2^(- (∆∆CT)) میزان بیان ژن هدف با ژن مرجع نرمالیز شده و بیان ژنهای گروه سالم بهعنوان کالیبراتور در نظر گرفته شد. مشخصات پرایمرهای سنتز شده در جدول 1 ذکرشده است.
تجزیهو تحلیل آماری
جهت تعیین نرمال بودن دادهها از آزمون کلوموگروف- اسمیرنوف Kolmogorov–Smirnov test استفاده شد. برای بررسی معنیدار بودن اختلاف بین گروهها از تحلیل واریانس یکطرفه و در صورت معنیداری، جهت تعیین تفاوت بین میانگینهای دوگروهی از آزمون تعقیبی توکی استفاده شد. تجزیه و تحلیل دادهها با استفاده از نرمافزارversion 16 SPSS در سطح معنیداری 0/5 (0/05>p) انجام شد.
ملاحظات اخلاقی
پروپوزال این تحقیق توسط دانشگاه لرستان با کد اخلاق (IR.SSRC.REC.1397.017) تایید شده است
نتایج
نتایج نشان داد که وزن اولیه گروهها اختلاف معناداری با یکدیگر نداشت (0/328=P)، اما در هفتههای پایانی پژوهش، میانگین تغییرات وزن موشهای گروههای نوروپاتی دیابت نسبت به گروه شاهد بهصورت معناداری کمتر بود (0/001=P). همچنین، میانگین تغییرات وزن گروه نوروپاتی دیابت تمرین نسبت به گروه نوروپاتی دیابت، در هفته هشتم افزایش معنادار داشتند (0/063=P) (جدول 2). پس از القاء دیابت، سطوح گلوکز خون بهصورت معناداری در گروههای نوروپاتی دیابت افزایش یافت (0/001=P) و این اختلاف تا پایان دوره پژوهش در مقایسه با گروه شاهد همچنان معنیدار بود (0/001=P)، همچنین، در پایان برنامه تمرینی، غلظت گلوکز خون گروه نوروپاتی دیابت تمرین نسبت به گروه نوروپاتی دیابت بهصورت معناداری پایینتر بود (0/001=P) ولی بااینحال همچنان اختلاف معنیداری با گروه شاهد وجود داشت (0/012=P) (جدول 2). میانگین مدتزمان تأخیر در پس کشیدن پنجه (Paw withdrawal latency) در آزمون هات پلیت دو هفته پس از القاء دیابت در گروههای نوروپاتی دیابت نسبت به گروه شاهد بهطور معنیداری کمتر بود (0/001=P). همچنین در هفتههای پایانی اجرای پروتکل تمرین هوازی، میانگین مدتزمان تأخیر در پس کشیدن پنجه در آزمون هایپرآلژزیای حرارتی در گروه نوروپاتی دیابت تمرین نسبت به گروه نوروپاتی دیابت بهطور معنیداری بیشتر بود (0/001=P) (نمودار 1).
دو هفته بعد از القاء دیابت، میانگین تغییرات آستانه پس کشیدن پنجه در آزمون آلودینیای مکانیکی در گروه¬های نوروپاتی دیابت نسبت به گروه شاهد بهطور معنی¬داری کمتر بود (0/005=P). از طرفی در هفتههای هفتم و هشتم اجرای پروتکل، میانگین تغییرات آزمون آلودینیای مکانیکی در گروه نوروپاتی دیابت تمرین افزایش معنیداری نسبت به گروه نوروپاتی دیابت داشتند (026/0=P) (نمودار2). با توجه به میانگین گروهها، مشخص شد که القاء دیابت موجب کاهش معنادار در میزان بیان ژن IL-35 و افزایش معناداری در ژن NF-kB در بخش خلفی نخاع موشهای نوروپاتی دیابت در مقایسه با گروه شاهد شد (0/001=P) (P=0/021). میزان بیان ژن IL-35 در گروه نوروپاتی دیابت تمرین بهطور معناداری نسبت به گروه نوروپاتی دیابت بالاتر بود (0/007=P). همچنین میزان بیان ژن NF-kB در گروه نوروپاتی دیابتی تمرین بهطور معناداری نسبت به گروه نوروپاتی دیابت پائینتر بود (0/034=P). از طرفی میزان بیان ژنهای فوق بین گروهای نوروپاتی دیابت تمرین و گروه شاهد تفاوت معنیدار نشان داده نشد (0/2=P) (نمودار 3).
جدول 1: مشخصات توالی پرایمر های ژنهای مورداستفاده در پژوهش
جدول 2: نتایج آزمون تحلیل واریانس دو طرفه وزن بدن و سطح گلوکز خون در موشهای گروههای مختلف
کلیه مقادیر جدول بهصورت انحراف استاندارد ± میانگین میباشند. *اختلاف معنیدار با گروه شاهد (0/05>p). †اختلاف معنیدار با گروه نوروپاتی دیابتی (0/05>p).
نمودار 1: تغییرات زمان تأخیر در پس کشیدن پنجه در آزمون هایپرآلژزیای حرارتی گروههای مختلف
* اختلاف معنیدار با گروه شاهد P<0.05)). † اختلاف معنیدار با گروه نوروپاتی دیابتی P<0.05)).
نمودار 2: تغییرات آستانه پس کشیدن پنجه در آزمون آلودینیای مکانیکی گروههای مختلف
* اختلاف معنیدار با گروه شاهد P<0.05)). † اختلاف معنیدار با گروه نوروپاتی دیابتی P<0.05)).
نمودار 3: میزان بیان ژن NF-κB و IL-35 در بخش خلفی نخاع گروههای مختلف.
* اختلاف معنیدار با گروه شاهد P<0.05)). † اختلاف معنیدار با صگروه نوروپاتی دیابتی P<0.05)).
بحث
در پژوهش حاضر اثر تمرین هوازی بر میزان بیان ژن IL-35 و NF-kB در بافت نخاع موشهای مبتلابه درد نوروپاتی دیابتی و همچنین آستانه پاسخ به آزمونهای رفتاری درد نوروپاتی، آزمونهات پلیت (حساسیت سیستم عصبی به محرکهای دردناک) و آزمون آلودینیای مکانیکی (حساسیت سیستم عصبی به محرکهای بدون درد) مورد بررسی قرار گرفت. یافتههای پژوهش حاضر نشان داد که پس از القاء و اثبات درد نوروپاتی دیابت ناشی از تزریق استرپتوزوسین، تمرین هوازی، میزان بیان ژن IL-35 را در بخش خلفی نخاع بهطور معنیداری افزایش و میزان بیان ژن NF-kB را کاهش و آستانه پاسخ به آزمونهای رفتاری درد نوروپاتی را افزایش داده است. تحقیقات زیادی نشان دادهاند که اتو اکسیداسیون گلوکز به دنبال هیپرگلیسمی باعث تولید بیرویه رادیکالهای آزاد و سطوح عوامل التهابی در سلولها ازجمله نورونها میشود، گمان میرود که این تغییرات بیوشیمیایی در سلولهای گلیال و نورونهای محیطی باعث اختلال در حساسیت طبیعی نوسیسپتورها به محرکهای درد زا و غیر درد زا میشود (23). بهطورکلی پذیرفتهشده است که تولید بیشازحد واسطههای التهابی درنتیجه دیابت، نقش مهمی در شروع و حفظ درد نوروپاتی دیابتی دارد (24). در شرایط فیزیولوژی طبیعی، میکروگلیاها واسطههای ضدالتهابی را آزاد و بقایای سلولهای سمی را فاگوسیتوز کرده تا هموستاز را در سیستم عصبی حفظ کنند. مشخص شده است که میکروگلیاها توسط عوامل متعددی از جمله هایپرگلیسمی فعال میشوند، فعالسازی طولانیمدت میکروگلیاها باعث تولید بیشازحد سیتوکین های التهابی شده که این عامل منجر به التهاب عصبی میشود (25). پژوهشها نشان دادند که تحت شرایط استرس ناشی از هایپرگلیسمی، فنوتیپ سلولهای میکروگلیال به M1 تغییر کرده و میزان بیان ژنهای پیش التهابی IL-6، IL-1β و TNF-α افزایش مییابند، این عامل نقش مهمی در شروع و تداوم درد نوروپاتیک دیابتی دارد (26). تحقیقات اخیر گزارش دادهاند که عواملی میتوانند پلاریزاسیون میکروگلیالها از فنوتیپ M1 پیش التهابی به فنوتیپ ضدالتهابی M2 را تغییر دهند، شناسایی و بهکارگیری این عوامل اثرگذار در این تغییر نشاندهنده یک راهبرد درمانی جدید برای درد نوروپاتی دیابتی است. محققین افزایش سطوح عوامل التهابی CD68، iNOS، IL-6، IL-1β و TNF-α را بهعنوان نشانگرهای فعالیت فنوتیپ M1 میکروگلیالها عنوان کردهاند (9,27). نتایج حاصل از دادههای پژوهش حاضر نشان داد که میزان بیان ژن فاکتور رونویسی NF-kB، القاء کننده عوامل التهابی در گروه نوروپاتی دیابتی افزایش بیانشده بود. افزایش بیان این عامل رونویسی در نخاع، نشان بر فعالیت میکروگلیال ها فنوتیپ M1 دارد. آزمایشهای حیوانی و مطالعات In Vitro نشان دادهاند که IL-35 برونزا از طریق مهار مسیر پیامرسانی JNK و تنظیم مثبت مسیر سیگنالینگ JAK2/STAT6 نقش مهمی در پلاریزاسیون ماکروفاژها از فنوتیپ M1 التهابی به فنوتیپ ضدالتهابی M2 ایفا میکند (8,9). با توجه به اینکه افزایش نشانگرهای فعالیت میکروگلیال های فنوتیپ M2 شاخصهای ضدالتهابی همانند ARG-1، IL-10 و IL-35 هستند (9). احتمالاً کاهش میزان بیان ژن IL-35 در بافت نخاع موشهای صحرایی گروه نوروپاتی دیابتی در پژوهش حاضر، کاهش فعالیت فنوتیپ M2 را در این گروه نشان دهد. تحقیقات نشان دادهاند که سلولهای سیستم ایمنی اثر بیولوژیکی خود را از طریق تنظیم بیان سایتوکین های ضدالتهابی مانند IL-10، IL-4 و IL-35 انجام میدهند (28)؛ بنابراین هدف قرار دادن سایتوکین ضدالتهابی IL-35 و سنتز آن در نورونهای حسی، ممکن است، بهعنوان یک رویکرد مهم درمانی برای درد نوروپاتیک دیابت استفاده شود. مطالعات اثرات محافظتی تمرین هوازی را در طی شرایط فیزیولوژیکی و پاتولوژیکی ازجمله تنظیم سیستم ایمنی، افزایش سطوح شاخصهای ضدالتهابی و افزایش ظرفیت آنتیاکسیدانی بهخوبی به اثبات رساندهاند (29). ولی هیچگونه مطالعهای، نقش فعالیت هوازی را بر سایتوکین ضدالتهابی IL-35 در سیستم عصبی محیطی و رابطه آن با آزمونهای رفتاری درد نورپاتیک انجام نگرفته است. مطالعات نشان دادهاند که فعالیتهای ورزشی قطبش میکروگلیال های نخاع را به فنوتیپ M2 تغییر داده و نوروپاتی دیابتی را بهبود بخشیده است (30)؛ بنابراین این احتمال وجود دارد که فعالیت هوازی، بهواسطه اثرات آنتیاکسیدانی و ضدالتهابی، توانسته است تولید پروتئینهای آنتیاکسیدان و عوامل ضدالتهابی مانند IL-35 را تنظیم مثبت کرده و از این طریق با تأثیر و دخالت بر مهار مسیرهای التهابی در سیستم عصبی، باعث کاهش عوارض نوروپاتی دیابتی در موشهای صحرایی مورد آزمون شده باشد. بااینحال، عدم اندازهگیری پروتئینهای دفاعی آنتیاکسیدانهای آنزیمی و غیر آنزیمی که در نتیجه نوروپاتی دیابتی تنظیم مثبت میشوند، در پژوهش حاضر بهعنوان یک محدودیت از بحث دقیق در این زمینه جلوگیری میکند. با این وجود پیشنهاد میشود در پژوهشهای آتی، همراه با اندازه¬گیری سایتوکین ضدالتهابی IL-35 سایر عوامل ضدالتهابی مانند IL-10 و IL-4 بهعنوان شاخصهای فعالیت میکروگلیاهای فنوتیپ M2 و مارکرهای فنوتیپ M1 شامل CD68، iNOS، IL-6، IL-1β و TNF-α برای دستیابی به شرایط قطعیتر مورد بررسی قرار دهند.
نتیجه گیری
پژوهش حاضر نشان داد که القاء دیابت باعث کاهش میزان بیان ژن IL-35 و افزایش بیان NF-kB در بخش حسی نخاع شده و این عامل منجر به افزایش حساسیت گیرندههای درد میشود. بااینحال شش هفته تمرین هوازی توانسته است این میزان بیان سایتوکین ضدالتهابی IL-35 را تنظیم مثبت و بیان فاکتور رونویسی NF-kB را تنظیم منفی کند و حساسیت سیستم عصبی به محرکهای دردناک را کاهش دهد. لذا بهنظر میرسد که تمرین هوازی از طریق افزایش سطوح سایتوکینهای ضدالتهابی منجر به کاهش التهاب عصبی شده و از این طریق در کاهش عوارض نوروپاتی دیابتی اثرگذار بوده است.
سپاسگزاری
این مقاله مستخرج از پایاننامه دورۀ دکتری رشته فیزیولوژی ورزشی میباشد؛ از کلیه کارشناسان محترم آزمایشگاه و اساتید گروه فیزیولوژی ورزشی دانشگاه لرستان تشکر و قدردانی میشود. ضمناً کلیه هزینههای این طرح شخصی تأمینشده است.
حامی مالی: ندارد.
تعارض منافع: وجود ندارد.
References:
1- Feldman EL, Nave KA, Jensen TS, Bennett DL. New Horizons in Diabetic Neuropathy: Mechanisms, Bioenergetics, and Pain. Neuron 2017; 93(6): 1296-313.
2- Calcutt NA. Diabetic Neuropathy and Neuropathic Pain: A (Con) Fusion of Pathogenic Mechanisms? Pain 2020; 161(Suppl 1): S65-8.
3- Schreiber AK, Nones CF, Reis RC, Chichorro JG, Cunha JM. Diabetic Neuropathic Pain: Physiopathology and Treatment. World J of diabetes 2015; 6(3): 432-44.
4- Sai Laxmi M, Prabhakar O. Inflammatory Biomarkers as a Part of Diagnosis in Diabetic Peripheral Neuropathy. Journal of Diabetes and Metabolic Disorders 2021; 20: 869-82.
5- Pop-Busui R, Ang L, Holmes C, Gallagher K, Feldman EL. Inflammation as a Therapeutic Target for Diabetic Neuropathies. Curr Diab Rep 2016; 16(3): 29.
6- Li X, Mai J, Virtue A, Yin Y, Gong R, Sha X, et al. IL-35 is a Novel Responsive Anti-Inflammatory Cytokine—a New System of Categorizing Anti-Inflammatory Cytokines. PloS One 2012; 7(3): e33628.
7- Yang XF, Li X, Mai J, Virtue A, Yin Y, Sha X, et al. IL-35 is a Novel Responsive Anti-Inflammatory Cytokine-A New System of Categorizing Anti-Inflammatory Cytokines. American Society of Hematology; 2012; 120(21): 5197.
8- Jiang Y, Wang J, Li H, Xia L. IL-35 Alleviates Inflammation Progression in a Rat Model of Diabetic Neuropathic Pain Via Inhibition of JNK Signaling. J Inflamm (Lond) 2019; 16: 19.
9- Jiang Y, Wang J, Li H, Xia L. IL-35 Promotes Microglial M2 Polarization in a Rat Model of Diabetic Neuropathic Pain. Arch Biochem Biophys 2020; 685: 108330.
10- Hosseini A, Abdollahi M. Diabetic Neuropathy and Oxidative Stress: Therapeutic Perspectives. Oxid Med Cell Longev 2013; 2013: 168039.
11- Singleton JR, Smith AG, Marcus RL. Exercise as Therapy for Diabetic and Prediabetic Neuropathy. Curr Diab Rep 2015; 15(12): 120.
12- Yan J-E, Yuan W, Lou X, Zhu T. Streptozotocin-Induced Diabetic Hyperalgesia in Rats is Associated with Upregulation of Toll-Like Receptor 4 Expression. Neurosci lett 2012; 526(1): 54-8.
13- Morrow TJ. Animal Models of Painful Diabetic Neuropathy: the STZ Rat Model. Current Protocols in Neuroscience 2004; Chapter 9: Unit 9.18.
14- Wei M, Ong L, Smith MT, Ross FB, Schmid K, Hoey AJ, et al. The Streptozotocin‐Diabetic Rat as a Model of the Chronic Complications of Human Diabetes. Heart Lung Circ 2003; 12(1): 44-50.
15- Malmberg AB, Bannon AW. Models of Nociception: Hot‐Plate, Tail‐Flick, and Formalin Tests in Rodents. Current Protocols in Neuroscience 1999; 6(1): 8.9. 1-8.9. 15.
16- Chen YW, Hsieh PL, Chen YC, Hung CH, Cheng JT. Physical Exercise Induces Excess Hsp72 Expression and Delays the Development of Hyperalgesia and Allodynia in Painful Diabetic Neuropathy Rats. Anesthesia & Analgesia 2013; 116(2): 482-90.
17- Yoon H, Thakur V, Isham D, Fayad M, Chattopadhyay M. Moderate Exercise Training Attenuates Inflammatory Mediators in DRG of Type 1 Diabetic Rats. Experimental Neurology 2015; 267: 107-14.
18- Rossi DM, Valenti VE, Navega MT. Exercise Training Attenuates Acute Hyperalgesia in Streptozotocin-Induced Diabetic Female Rats. Clinics (Sao Paulo) 2011; 66(9): 1615-9.
19- Gong YH, Yu XR, Liu HL, Yang N, Zuo PP, Huang YG. Antinociceptive Effects of Combination of Tramadol and Acetaminophen on Painful Diabetic Neuropathy in Streptozotocin-Induced Diabetic Rats. Acta Anaesthesiol Taiwan 2011; 49(1): 16-20.
20- Chae CH, Jung SL, An SH, Jung CK, Nam SN, Kim HT. Treadmill Exercise Suppresses Muscle Cell Apoptosis by Increasing Nerve Growth Factor Levels and Stimulating P-Phosphatidylinositol 3-Kinase Activation in the Soleus of Diabetic Rats. J Physiol Biochem 2011; 67(2): 235-41.
21- Gelderd JB, Chopin SF. The Vertebral Level of Origin of Spinal Nerves in the Rat. Anat Rec 1977; 188(1): 45-7.
22- Keri KC, Samji NS, Blumenthal S. Diabetic Nephropathy: Newer Therapeutic Perspectives. J Community Hosp Intern Med Perspect 2018; 8(4): 200-7.
23- Thakur V, Sadanandan J, Chattopadhyay M. High-Mobility Group Box 1 Protein Signaling in Painful Diabetic Neuropathy. Int J Mol Sci 2020; 21(3): 881.
24- Ismail CAN, Suppian R, Ab Aziz CB, Long I. Expressions of Spinal Microglia Activation, BDNF, and DREAM Proteins Correlated with Formalin-Induced Nociceptive Responses in Painful and Painless Diabetic Neuropathy Rats. Neuropeptides 2020; 79: 102003.
25- Wang D, Couture R, Hong Y. Activated Microglia in the Spinal Cord Underlies Diabetic Neuropathic Pain. Eur J Pharmacol 2014; 728: 59-66.
26- Jin GL, Hong LM, Liu HP, Yue RC, Shen ZC, Yang J, et al. Koumine Modulates Spinal Microglial M1 Polarization and the Inflammatory Response through the Notch-RBP-Jκ Signaling Pathway, Ameliorating Diabetic Neuropathic Pain in Rats. Phytomedicine 2021; 90: 153640.
27- Xiaohua G, Dongdong L, Xiaoting N, Shuoping C, Feixia S, Huajun Y, et al. Severe Vitamin D Deficiency is Associated with Increased Expression of Inflammatory Cytokines in Painful Diabetic Peripheral Neuropathy. Front Nutr 2021; 8: 612068.
28- da Luz Scheffer D, Latini A. Exercise-Induced Immune System Response: Anti-Inflammatory Status on Peripheral and Central Organs. Biochim Biophys Acta Mol Basis Disease 2020; 1866(10): 165823.
29- Sun JS, Yang YJ, Zhang YZ, Huang W, Li ZS, Zhang Y. Minocycline Attenuates Pain by Inhibiting Spinal Microglia Activation in Diabetic Rats. Mol Med Rep 2015; 12(2): 2677-82.
30- Gong X, Chen Y, Fu B, Jiang J, Zhang M. Infant Nerve Injury Induces Delayed Microglial Polarization to The M1 Phenotype, and Exercise Reduces Delayed Neuropathic Pain By Modulating Microglial Activity. Neuroscience 2017; 349: 76-86.
نوع مطالعه: پژوهشي |
موضوع مقاله:
فیزیولوژی ورزش
دریافت: 1401/3/16 | پذیرش: 1401/3/28 | انتشار: 1402/1/15
دریافت: 1401/3/16 | پذیرش: 1401/3/28 | انتشار: 1402/1/15
ارسال پیام به نویسنده مسئول
| بازنشر اطلاعات | |
 |
این مقاله تحت شرایط Creative Commons Attribution-NonCommercial 4.0 International License قابل بازنشر است. |
