

دوره 29، شماره 9 - ( آذر 1400 )
جلد 29 شماره 9 صفحات 4095-4083 |
برگشت به فهرست نسخه ها


![]()
![]()
![]()
Download citation:
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:



BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
Barati Z, Yaghoubi A, Jalilvand M R. Effect of Continuous and Interval Training on Amyloid β 42 (Aβ42) and Malondialdehyde (MDA) Levels in Hippocampus of Elderly Rats. JSSU 2021; 29 (9) :4083-4095
URL: http://jssu.ssu.ac.ir/article-1-5383-fa.html
URL: http://jssu.ssu.ac.ir/article-1-5383-fa.html
براتی زهرا، یعقوبی علی، جلیلوند محمدرضا. تأثیر تمرینات تداومی و تناوبی بر سطح آمیلوئید بتا 42 (Aβ42) و مالون دی آلدئید (MDA) هیپوکامپ موش های صحرایی نر سالمند. مجله علمي پژوهشي دانشگاه علوم پزشكي شهید صدوقی يزد. 1400; 29 (9) :4083-4095



واژههای کلیدی: تمرین تداومی، تمرین تناوبی، آمیلوئید بتا 42 (Aβ42)، مالون دی آلدئید (MDA)، موش های سالمند
متن کامل [PDF 1075 kb]
(1088 دریافت)
| چکیده (HTML) (2902 مشاهده)
متن کامل: (3340 مشاهده)
مقدمه
همانند سایر سیستمهای بدن، تواناییهای عملکردی مغز در طی پیری به تدریج کاهش مییابد، که بهصورت کاهش در یادگیری و حافظه، توجه، سرعت تصمیمگیری، درک حسی (بینایی، شنوایی، لمس، بو و چشایی) و هماهنگی حرکتی آشکار میشود (1). یکی از مهمترین تغییرات ناشی از افزایش سن، کاهش عملکرد شناختی میباشد (2). روند زمانی کاهش عملکرد مغزی وابسته به سن، تقریباً به موازات کاهش عملکرد سایر سیستمهای بدن با شتاب قابلتوجهی، بعداز 50 سالگی، افزایش مییابد (3). بیماری آلزایمر بهعنوان شایعترین بیماری تحلیل برنده عصبی وابسته به سن میباشد؛ مطالعات جهانی از افزایش روز افزون این بیماری در میان جمعیت سالمند در جوامع مختلف دنیا نشان دارد و گفته میشود در حال حاضر 36 میلیون نفر مبتلا به آلزایمر در سراسر جهان هستند. پیشبینیها حاکی است که باتوجه به افزایش جمعیت سالمند در جهان، آمار مبتلایان به آلزایمر تا سال 2050 به 115 میلیون نفر خواهد رسید (4). یکی از عوامل اصلی ایجادکننده آلزایمر، تشکیل پلاکهای پیری متشکل از پپتید آمیلوئید بتا (Amyloid Beta) میباشد (5). آمیلوئید بتا بهصورت طبیعی در مقادیر اندک در مغز یافت میشود و دارای 49-37 اسید آمینه میباشد که در اثر پروتئولیز APP ایجاد میشود (6، 5). بیشترین آمیلوئید بتای ترشح شده، حاوی 40 اسید آمینه (Aβ40) است و درصد اندکی، حاوی 42 اسید آمینه (Aβ42) میباشد. آمیلوئید بتای 42، به دلیل حضور 2 اسید آمینه آب دوست بیشتر در ترکیبش، نسبت به Aβ40 آسانتر تجمع مییابد و سمیت بیشتری دارد (7). اشاره شده است که در یک مغز طبیعی Aβ40، حدود 90 درصد و Aβ42 تقریباً 10-5 درصد، پپتیدهای Aβ را تشکیل میدهند. پپتید Aβ42 اولین و اصلیترین پپتید برای تشکیل پلاکهای آمیلوئیدی میباشد. بنابراین اندازهگیری سطوح Aβ42 و نسبت Aβ42/Aβ40 به عنوان یک روش شناسایی افراد مستعد و مبتلا به آلزایمر مورد استفاده قرار میگیرد (8). علاوه بر این sojkova و همکاران (2011) در یک مطالعه طولی به مدت 1/5 سال نشان دادند که رسوب و پلاکهای Aβ مغز بهصورت وابسته به سن، در طی دوره تحقیق، حتی در سالمندان بدون علامت آلزایمر نیز افزایش مییابد (9). از طرفی نشان داده شده است که افزایش شاخصهای استرس اکسایشی در آلزایمر با تجمع و رسوب Aβ در مغز همراه است (10). آمیلوئید بتا در وضعیتهای تجمعی مختلفی مشاهده میشود که در این بین Aβ الیگومریزه شده بهعنوان سمی¬ترین شکل در نظر گرفته می-شود (11). Aβ الیگومریزه میتواند در میتوکندری نیز یافت شود (12) که مهمترین منبع تولید رادیکالهای آزاد میباشد. مطالعات در شرایط آزمایشگاهی نشان داده است که تزریق Aβ42 به نورونهای اولیه هیپوکامپی در محیط کاشت منجر به افزایش شاخصهای استرس اکسایشی و سمیت عصبی میشود (14،13). همراه با این استرس اکسایشی ناشی از پپتید Aβ، افزودن ویتامین E بهعنوان یک آنتی اکسیدان، بهطور معناداری استرس اکسایشی و اثرات سمیت عصبی ناشی از Aβ42 را تعدیل میکند (13) که این موضوع پیشنهاد میکند که سمیت عصبی ناشی از Aβ42 از طریق توانایی این پپتید سمی در ایجاد استرس اکسایشی، میانجیگری میشود. در این بین Casado و همکاران (2008) نشان دادند که سطح مالون دی آلدئید (Malondialdehyde) به عنوان شاخص پراکسیداسیون لیپیدی، همراه با افزایش سن در بیماران مبتلا به آلزایمر افزایش مییابد و استرس اکسایشی نقش مهمی در آسیبهای مغزی بیماران آلزایمری بازی میکند (15). شناسایی عواملی که سطح Aβ42 را در مغز تنظیم میکنند، هدف مهمی برای افزایش عملکرد و سلامت مغز میباشد (16). تمرین و فعالیت ورزشی یکی از راه کارهای حمایتی و غیرتهاجمی برای بهبود عملکرد مغز میباشد. یک مطالعه موردی نشان داده است که افراد مبتلا به اختلالات حافظه و یادگیری، در میانسالی کمتر فعال بودهاند و این غیرفعال بودن با 25 درصد افزایش توسعه آلزایمر همراه میباشد (17). به طور مشابه، تحقیق دیگری نشان داده است که فعالیت بدنی در میانسالی در برابر توسعه اختلالات شناختی، بیماری آلزایمر و جنون محافظت ایجاد میکند و فعالیت بدنی 60 درصد کاهش در شیوع آلزایمر در سالمندی را به همراه دارد (18). Yu و همکاران (2013) نشان دادند که تمرین ورزشی از طریق کاهش معنادار در سطح Aβ42، کاهش سطح MDA و بهبود فعالیت سوپراکساید دیسموتاز (Superoxide dismutase)، را در پی داشته است که با بهبود حافظه و سلامت مغز همراه است (19). Zhao و همکاران (2015) نشان دادند که 5 ماه تمرین ورزشی نوارگردان از تجمع Aβ (در سن 17 ماهگی) جلوگیری میکند و بنابراین میتوان از تمرین نوارگردان برای کند کردن پیشرفت آلزایمر بعد از مرحله تجمع پلاکهای آمیلوئیدی استفاده کرد (20). همچنین یعقوبی و همکاران (1395) عنوان داشتند که تمرین ورزشی تداومی باعث کاهش Aβ42 هیپوکامپ موشهای آلزایمری میشود (21). تحقیقات مختلفی به بررسی سازوکارهای درگیر در تعدیل سطح Aβ42 متعاقب فعالیت ورزشی با شدت پایین و متوسط را مورد بررسی قرار دادهاند (23، 22) ولی اثر پروتکل های تمرین طولانیمدت و سازوکارهای احتمالی ناشی از آن به خوبی درک نشده است. در نتیجه نتایج تحقیق حاضر برای یافتن استراتژیهای بالقوه و طراحی تمرین و یافتن پروتکل تمرین مناسب برای پیشگیری از انحطاط عصبی ناشی از سالمندی مفید خواهد بود. بنابراین هدف از تحقیق حاضر بررسی اثر تمرینات تداومی و تناوبی بر سطح Aβ42 و MDA در هیپوکامپ موشهای صحرایی نر سالمند بود.
روش بررسی
پژوهش حاضر از نوع تجربی با طرح پس آزمون به همراه گروه کنترل بود که به شیوه آزمایشگاهی انجام شد. در این تحقیق از 30 سر موش سالمندنر نژاد ویستار با دامنه وزنی 320 تا 380 گرم و سن 18 ماهه (24) استفاده شد که از آزمایشگاه حیوانی دانشگاه آزاد اسلامی واحد مرودشت خریداری گردید. مطابق با خطمشی انجمن ایرانیان حمایت از حیوانات آزمایشگاهی در قفسهای 4تایی و تحت شرایط استاندارد (چرخه 12 ساعته روشنایی- تاریکی، دمای 2±25 درجه سانتیگراد) با دسترسی آزاد به آب و غذا نگهداری شدند. پس از یک هفته آشنایی و سازگاری با محیط جدید، موشهای صحرایی نر سالمند بهطور تصادفی و بر اساس وزن به سه گروه تمرین تناوبی، تمرین تداومی و کنترل با تعداد برابر در هر گروه (10 سر) تقسیم شدند. در ادامه، موشهای نر سالمند با راه رفتن و دویدن بر روی نوارگردان آشنا شدند. پس از یک هفته آشناسازی، حداکثر سرعت دویدن با استفاده از آزمون عملکرد ورزشی مدرج برآورد شد. آنگاه، موشهای گروههای تجربی در هشت هفته تمرینات تناوبی یا تمرین تداومی دویدن روی نوارگردان شرکت کردند. 48 ساعت پس از آخرین جلسه تمرینی و پس از ناشتایی شبانه، تمامی موشهای بیهوش شده و بافتبرداری صورت گرفت. لازم به ذکر است تمامی موشهای در طول مداخله رژیم غذایی استاندارد را مصرف کردند. تمامی مداخلات حیوانی مطابق با دستورالعملهای اخلاقی مؤسسات ملی برای مراقبت و استفاده از حیوانات آزمایشگاهی انجام شد. پروتکل تمرین تداومی به مدت هشت هفته و 5 جلسه در هفته بر روی نوارگردان (شیب صفر درجه) اجرا شد. پروتکل تمرین تداومی در 3 مرحله آشنایی، اضافه بار و تثبیت بار اجرا شد. در مرحله آشنایی (هفته اول)، موشها هر روز به مدت 15-10 دقیقه با سرعت 10 متر در دقیقه روی نوارگردان راه رفتند. در مرحله اضافه بار (هفته دوم تا پنجم)، به تدریج در طی 3 هفته، به شدت و مدت فعالیت افزوده شد؛ تا به میزان نهایی 40 دقیقه با سرعت 20 متر در دقیقه رسید. در مرحله حفظ یا تثبیت (هفته ششم تا هشتم)، تمرین با همین شدت ادامه یافت تا 8 هفته به پایان رسید. این پروتکل با شدت 65 تا 70% VO2max موشهای سالمند بود. همچنین، 10 دقیقه گرمکردن و 5 دقیقه سردکردن با شدت پایین در ابتدا و انتهای هر جلسه تمرینی اجرا شد (25). پروتکل تمرین تناوبی نیز به مدت هشت هفته و 5 جلسه در هفته بر روی نوارگردان (شیب صفر درجه) اجرا شد. این پروتکل، شامل اجرای 8-5 مرحله فعالیت 2 دقیقهای با شدت معادل 80 تا 100 درصد اکسیژن مصرفی بیشینه و با دورههای استراحتی فعال 2 دقیقهای با شدت معادل 50 درصد اکسیژن مصرفی بیشینه بود. بر این اساس، سرعت دویدن در هفته اول از 22 متر بر دقیقه به 31 متر بر دقیقه در دو هفته پایانی رسید. همچنین، تعداد تکرار تناوب در هفته اول از 5 تکرار به 8 تکرار در هفته چهارم رسید و در ادامه نیز با 8 تکرار اجرا شد. همچنین، 10 دقیقه گرمکردن و 5 دقیقه سردکردن با شدت پایین در قبل و بعد از هر جلسه تمرینی در نظر گرفته شد (27-25). تمامی آزمودنیها، 72 ساعت پس از آخرین جلسه تمرین، با تزریق درون صفاقی ترکیبی از کتامین (50 میلیگرم به ازای هر کیلوگرم) و زایلازین (4 میلیگرم به ازای هر کیلوگرم) بیهوش شدند. برای جمعآوری نمونههای هیپوکامپ، سر آزمودنی ها از ناحیه گردن توسط قیچی مخصوص جدا شد، ابتدا با استفاده از تیغ جراحی، جمجمه شکافته شده و مغز با احتیاط خارج گردید. مغز سالم توسط تیغ جراحی دقیقاً از وسط به دو نیم تقسیم شد و با توجه به مختصات هیپوکامپ به کمک اطلس پاک سینوس، هیپوکامپ از سیستم لمبیک جدا شد. نمونههای هیپوکامپ جمعآوری شده برای اندازهگیریهای بعدی در دمای ۸۰- درجه سانتیگراد نگهداری شد. برای اندازهگیری مقادیر پروتئینی Aβ42 و MDA از روش وسترن بلات استفاده شد. برای استخراج پروتئین های هیپوکامپ از بافر سنجش رسوب رادیوایمنی (Radioimmunoprecipitation Assay Buffer) حاوی 0/05 میلیمولار بافر تریس ( pHبرابر 8)، 150 میلیمولار کلرید سدیم، 0/01 درصد اتیلن گلیکول تترااسید استیک (Ethylene Glycol Tetra Acetic Acid))، یک درصد SDSبه اضافه 0/1 درصد آنتی پروتئاز کوکتیل (Protease Inhibitor Cocktail) استفاده شد. به این ترتیب که 100 میلیگرم بافت در 500 میکرولیتر بافر حاوی آنتی پروتئاز توسط یک هموژنایزر دستی هموژن شد و نیم ساعت در دمای 4 درجه سانتیگراد گذاشته شدند و سپس دریک سانتریفوژ یخچال دار (bo ,sw14rfroil) در دور 12000 و 4 درجه سانتیگراد و به مدت 10 دقیقه سانتریفوژ شد. مایع رویی جمعآوری شده و غلظت پروتئین آن با کیت تعیینکننده پروتئین (Bio-Rad) در طول موج 595 نانومتر اندازهگیری و در نهایت در دمای 20 درجه زیر صفر در فریزر نگهداری شد. سپس، هموژن به دست آمده به نسبت 1:1 با نمونه لودینگ بافر (mM 50تریس-کلرید هیدروژن، 2 درصد سدیم دو دسیل سولفات، 10 درصد گلیسرول، 5 درصد بتا- مرکاپتواتانول و 0/005 درصد برموفنول آبی) مخلوط گردید. آنگاه، نمونهها به مدت 5 دقیقه جوشانده شد تا تمام پروتئینها کاملاً دناتوره شوند. پروتئینها با استفاده از الکتروفورز ژل SDS-polyacrylamide جدا شده و به غشای نیتروسلولز منتقل شدند. غشا به مدت 1 ساعت در 5 درصد BSA در Tris-Buffered Saline و 0/1 درصد Tween 20 TBST)) مسدود شد و در آنتیبادی اولیه (1: 500) انکوبه شد. انکوباسیون در آنتیبادی ثانویه روز بعد به مدت 1 ساعت در دمای اتاق در 4 درصد TBSTانجام شد. پروتئینها با یک واکنش شیمیایی لومینسانس و با تجزیه و تحلیل densitometry با نرمافزار Image J اندازهگیری شدند. آنتیبادیهای اولیه و ثانویه Aβ42 (F-4):sc-390904 و mouse anti-rabbit IgG-HRP:sc-2357 از شرکتSanta Cruz Biotechnology, Inc مورد استفاده قرار گرفتند. سطح MDA به عنوان شاخصی از پراکسیداسیون لیپیدی در هیپوکامپ موشهای سالمند با روش جذب سنجی با استفاده از کیت شرکت زلبایو ساخت کشور آلمان در دامنه 532 (540-530) نانومتر اندازهگیری شد. بر اساس مقایسه با نمودار استاندارد نتایج حاصل، میزان MDA بر حسب نانو مول بر میلیگرم پروتئین بیان شد.
تجزیه و تحلیل آماری
از آزمون شاپیروولیک جهت بررسی توزیع طبیعی دادهها استفاده شد. جهت تعیین معنادار بودن تفاوت میانگین پس آزمون متغیرها بین گروههای تحقیق، از آزمون آنالیز واریانس یک طرفه استفاده گردید. اطلاعات مورد نیاز پس از جمعآوری، توسط نرمافزار آماری version 16 SPSS در سطح معناداری p< 0/05 مورد تجزیه و تحلیل قرار گرفت.
ملاحظات اخلاقی
پروپوزال این تحقیق توسط دانشگاه آزاد اسلامی واحد بجنورد تایید شده است (کد اخلاق IR.IAU.BOJNOURD.REC.1399.022).
نتایج
در این بخش اطلاعات توصیفی مربوط به وزن موشها در ابتدا و انتهای دوره پژوهش ارائه شده است. جهت اطمینان از همگن بودن گروهها از نظر وزن در پیش آزمون از آزمون آنالیز واریانس یک طرفه استفاده شد که نتایج آن در یک ستون در جدول 1 قرار داده شده است. تعداد نمونهها در تمام گروهها 10 سر بود که در طی دوره پژوهش با ریزش در گروه تمرین تناوبی و کنترل همراه بود که در نهایت در هر گروه 9 سر باقی ماندند ولی در موشهای تعداد گروه تمرین تداومی تغییری ایجاد نشد. با عنایت به جدول فوق و سطح معناداری گزارش شده میتوان دریافت گروهها در گروه بندی اولیه و انتهای دوره تحقیق تفاوت معناداری با یکدیگر نداشتند (0/05<p). به منظور مقایسه سطوح پروتئینی Aβ42 و MDA هیپوکامپ در گروههای تحقیق از آزمون آنووا (ANOVA) و آزمون تعقیبی توکی استفاده شد. در جدول 2 یافتههای آزمون آماری در خصوص مقایسه اثر تمرین تمرین تداومی و تمرین تناوبی بر سطح Aβ42 و MDA هیپوکامپ موشهای صحرایینر سالمند در گروههای تحقیق ارائه شده است. سطوح پروتئینی Aβ42 هیپوکامپ گروههای تحقیق. بر اساس نتایج حاصل از آزمون تعقیبی توکی، سطح پروتئین Aβ42 هیپوکامپ در گروه تمرین تداومی بهطور معناداری نسبت به گروه کنترل پایینتر بود (0/001=p). همچنین سطح این شاخص در گروه تمرین تناوبی بهطور معناداری نسبت به گروه کنترل پایینتر بود (0/001=p). ولی بین سطح پروتئین Aβ42 هیپوکامپ در گروه تمرین تداومی و تمرین تناوبی تفاوت معناداری مشاهده نشد (0/502=p). سطوح پروتئینی Aβ42 هیپوکامپ گروه¬های تحقیق، در نمودار 1 ارائه شده است. سطوح MDA هیپوکامپ گروههای تحقیق. بر اساس نتایج حاصل از آزمون تعقیبی توکی، سطح MDAهیپوکامپ در گروه تمرین تداومی بهطور معناداری نسبت به گروه کنترل پایینتر بود (0/016=p). همچنین سطح این شاخص در گروه تمرین تناوبی بهطور معناداری نسبت به گروه کنترل پایینتر بود (0/046=p). ولی بین سطح MDA هیپوکامپ در گروه تمرین تداومی و تمرین تناوبی تفاوت معناداری مشاهده نشد (0/866=p). سطوح MDA هیپوکامپ گروه¬های تحقیق، در نمودار 2 ارائه شده است.
جدول 1: مقایسه وزن آزمودنیها در پیش آزمون و پس از 8 هفته تمرین تداومی و تناوبی در گروههای تحقیق
.JPG)
* جهت مقایسه گروه ها از آزمون آنالیز واریانس یک طرفه استفاده شد.
جدول 2: مقایسه سطوح Aβ42 و MDA در هیپوکامپ گروههای تحقیق و یافتههای آزمون آنالیز واریانس
.JPG)
* نشانه تفاوت معنادار بین گروههای تحقیق در سطح 0/05>p.
.jpg)
نمودار1: مقایسه سطوح پروتئینی Aβ42 هیپوکامپ گروههای تحقیق پس از 8 هفته تمرین تناوبی و تداومی.
*نشانه تفاوت معنی دار نسبت به گروه کنترل در سطح 0/05>p
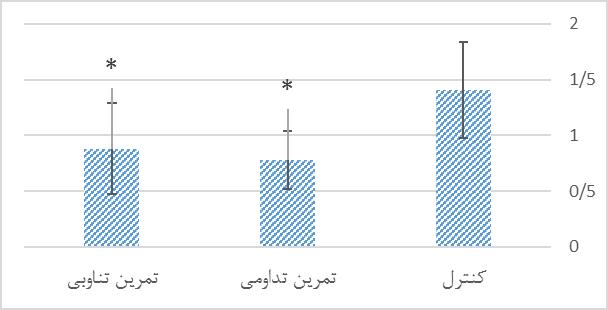
نمودار 2: مقایسه سطوح MDA هیپوکامپ گروههای تحقیق پس از 8 هفته تمرین تناوبی و تداومی.
*نشانه تفاوت معنی دار نسبت به گروه کنترل در سطح 0/05>p
بحث
نتایج تحقیق حاضر نشان داد که تمرین ورزشی فارغ از شدت و نوع تمرین سطح Aβ42 و MDA هیپوکامپ موشهای سالمند را کاهش داد. دو عامل اصلی بیماری آلزایمر شامل تشکیل پلاکهای پیری متشکل از پپتید Aβ و کلافههای نوروفیبریلار (Neurofibrillary tangle) متشکل از پروتئینهای هیپرفسفوریله تائو (Tau hyperphosphorylated) میباشد (5). ماهیت آب گریزی پپتیدهای Aβ42، به آنها اجازه میدهدکه تجمع یافته و پروتوفیبریلها و فیبریلها را تشکیل دهند و سرانجام تجمع این عوامل منجر به رسوب پلاک¬های آمیلوئیدی را می¬دهند (28). افزایش تولید Aβ42 و کاهش حذف آن از مغز عامل اصلی تجمع آن در مغز و آسیب به ارتباطات نرونی و کاهش حافظه و یادگیری بیان شده است (29). در این ارتباط بیان شده است که افزایش سطح و رسوب Aβ در پلاکهای خارج سلول بهطور ویژه باعث اختلال سیناپسی، اختلال در شبکه نورونی، اختلال در عملکرد میتوکندریایی، آپوپتوز سلول عصبی و کاهش حافظه میگردد (32-30). همچنین، هر دوی میکروگلیاها و آستروسیتها، پروتئین Aβ ترشح میکنند (30). پلاکهای پیری اصولاً متشکل از پپتیدهای Aβ می¬باشد که نقش اصلی آن در بیماریزایی آلزایمر به طور گستردهای ثابت شده است (33، 30). در حال حاضر شواهد وجود دارند مبنی بر اینکه تمرینات ورزشی، یک عامل مهم در برابر روند سالمندی و بیماریهای تحلیل برنده عصبی ناشی از آن میباشد (34). در مطالعه حاضر سطح Aβ42 هیپوکامپ موشها در هر دو تمرین تداومی و تناوبی نسبت به موش¬های گروه کنترل، بهطور معناداری پایینتر بود. نتایج مربوط به تأثیر تمرین تداومی و تناوبی نشان داد که سطح Aβ42 تفاوت معناداری بین دو پروتکل تمرینی وجود ندارد. این نتایج با نتایج Kang & Cho (2014) که نشان دادند 6 هفته تمرین نوارگردان از طریق بهبود سیگنال دهی انسولین، کاهش معنادار سطح Aβ42 را در مغز موش¬های آلزایمری را باعث شده است (35). همچنینLiu و همکاران (2013) نشان دادند که تمرین میتواند از طریق اثر مهاری بر سطح Aβ، به عنوان راهکار درمانی برای آلزایمر مطرح باشد (36). همچنین Kang و همکاران (2013) بیان داشتند که 12 هفته تمرین ورزشی روی نوارگردان از اختلال جهش در ژن PS2 جلوگیری کرده و تجمع Aβرا از طریق مهار فعالیت β-سکرتاز و فراورده های آن، کاهش داد (37). همچنین یعقوبی و همکاران (1394) با بررسی تأثیر تمرین تداومی با دو شدت پایین و بالا بر موشهای صحرایی آلزایمری شده عنوان داشتند که تمرین تداومی فارغ از شدت تمرین (پایین و بالا) باعث کاهش سطح Aβ42 هیپوکامپ موشهای آلزایمری شده میگردد (21). درباره تغییرات سطح Aβو متابولیسم آن در اثر تمرین، نظریههای مختلفی وجود دارد. با توجه به این که فعالیت ورزشی بسیاری از فرآوردههای ژنی را هم در سطح mRNA و هم در سطح پروتئین تعدیل می¬کند، القاءکننده تغییرات آناتومیکی، عصبیشیمیایی و الکتروفیزیولوژیکی افزایش دهنده شکلپذیری نورونی میباشد (38)، این احتمال وجود دارد که چندین مسیر برای تنظیم سطح Aβ بهطور مستقیم و یا به طور غیرمستقیم فعال باشد. به عنوان مثال یک روش احتمالی، افزایش فعالیت پروتئازوم (Proteasome) ناشی از ورزش است که قبلاً در عضله اسکلتی گزارش شده است (39) و می¬تواند تخریب قطعات پروتئولیتیکی APP را میانجیگریکند (40). اما Adlard و همکاران (2005) بیان داشتند که احتمالاً تمرین ورزشی می-تواند متابولیسم APP و آبشار Aβ در مغز را در راستای کاهش تولید Aβ میانجیگری کند که مستقل از سطح نپریلیزین و IDE به عنوان عوامل اصلی تجزیه Aβ42، میباشد (41). احتمال دوم این است که ورزش به طور مستقیم متابولیسم APP را با استفاده از افزایش فعالیت نورونی تعدیل میکند. به عنوان مثال، پردازش APP میتواند توسط پروتئینکیناز فعال¬شده با میتوژن (Mitogen-Activated Protein Kinases) و فسفولیپاز C تکمیل شود و ثابت شده است که این مسیرها از طریق ورزش فعال میشوند (42). از طرف دیگر، فعالیت کولینرژیک با ورزش افزایش مییابد و تنظیم سیستم کولینرژیک توسط ورزش در شکل¬پذیری نورونی ناشی از ورزش دخالت دارد (38). نقش MDA به عنوان یک شاخص از آسیب اکسایشی ناشی از بیماری و سالمندی به خوبی ثابت شده است (43). در مطالعه Gil و همکاران (2006) که 194 زن و مرد سالم بین سنین 18 تا 84 سال مورد بررسی قرار گرفتند، نشان داده شد که سطح MDA پلاسما با افزایش سن افزایش مییابد که نشان دهنده تسریع اکسیداسیون در طی سالمندی میباشد (44). نتایج محققان هندی که به بررسی 100 مرد سالم پرداختند نیز نشان میدهد که نسبت MDA/TAC (total antioxidant capacity) با افزایش سن افزایش می یابد (45). همچنین Dei و همکاران (2002) نشان دادند که سطح MDA در اطراف آستروسیتها، نورون ها و پلاکهای هیپوکامپی بیماران آلزایمری افزایش مییابد (46). دبیدی روشن و همکاران (2013)، در ارتباط با تأثیر تمرین ورزشی استقامتی بر استرس اکسایشی، بیان داشتند 8 هفته تمرین استقامتی باعث کاهش MDA به عنوان شاخص پراکسیداسیون لیپیدی در هیپوکامپ موشهای آلوده به سرب میشود (47). همچنین فلاح محمدی و همکاران (1390) نشان دادند که به دنبال اجرای هشت هفته تمرینات تداومی آثار تخریبی القائی توسط هموسیستئین در هیپوکامپ موشهای صحرایی کاهش یافته است که بهوسیله کاهش سطوح MDA و افزایش سطوح SOD مشخص شد (48). در زمینه ارتباط استرس اکسایشی و آسیبهای سیستم عصبی نیز ارتباط نزدیکی بین MDA و Aβ42 مورد بررسی قرار گرفته است و بهطور گستردهای ثابت شده است که تجمع پپتید Aβ در مغز باعث القای استرس اکسایشی و التهاب عصبی می¬گردد (33،30). شواهدی وجود دارد که Aβ42 داخل نورونی میتواند وارد میتوکندری شده، زنجیره انتقال الکترونی را مختل و تولید گونههای اکسیژن واکنش دهنده شامل رادیکالهای هیدروکسیل، سوپراکسید و پراکسیداسیون هیدروژن را القا کند (49). همچنین مطالعات در شرایط آزمایشگاهی نشان داده است که تزریق Aβ42 به نورونهای اولیه هیپوکامپی در محیط کاشت منجر به افزایش شاخصهای استرس اکسایشی و سمیت عصبی میشود (14،13). همراه با این استرس اکسایشی ناشی از پپتید Aβ، افزودن ویتامین E به عنوان یک آنتیاکسیدان، بهطور معناداری استرس اکسایشی و اثرات سمیت عصبی ناشی از Aβ42 را تعدیل میکند (13)، این موضوع پیشنهاد می¬کند که سمیت عصبی ناشی از Aβ42 از طریق توانایی این پپتید سمی در ایجاد استرس اکسایشی، میانجیگری میشود و بالعکس افزایش استرس اکسایشی و تخلیه آنتیاکسیدانی، افزایش سطح این شاخص را در مغز افراد آلزایمری در پی دارد. در این راستا تحقیق دیگری نشان داد که تخلیه ویتامین E، تجمع Aβ را از طریق کاهش پاکسازی از مغز و خون در موش¬های آلزایمری، افزایش میدهد (14). همچنین نشان داده شده است که تزریق Aβ40 به درون مغز موشها، با القای آسیب رادیکال آزاد و تغییرات در دفاع آنتیاکسیدانی مثل تخلیه گلوتاتیون در قشر پیشپیشانی و هیپوکامپ موش¬ها ارتباط دارد (50). علاوه بر این نشان داده شده است که مصرف مکملهای آنتی اکسیدانی و یا کاهش استرس اکسایشی کاهش سطح Aβ42 و بهبود عملکرد شناختی را در پی دارد. در این ارتباط 5 ماه خوراندن اسید گالیک و عصاره پلی¬فنولی دانه انگور غنی از کاتچین به موش-های آلزایمری، از بدتر شدن شناخت جلوگیری کرده و موجب کاهش سطوح اُلیگومرهای Aβ می¬شود (51). مکانیسم احتمالی موجود در این باب عبارت است از اینکه کاهش استرس اکسایشی و افزایش دفاع آنتیاکسیدانی به عنوان عامل افزایش دهنده فعالیت آنزیم α-سکرتاز و بازدارنده β- و γ-سکرتاز عمل می-کنند و بنابراین پردازش APPرا به سمت مسیر غیرآمیلوئیدوژنیک هدایت میکنند (52). از طرف دیگر بیان شده است که فعالیت ورزشی منظم ظرفیت آنتیاکسیدانی را بهبود می¬بخشد که در نتیجه منجر به جلوگیری از استرس اکسایشی، آپوپتوز و آسیب سلولی می¬شود و در مجموع احتمال خطر زوال عقل یا آلزایمر را کاهش میدهد (54، 53). در این راستا Um و همکاران (2008) نشان دادند که سطح SOD-1 و پروتئین کاتالاز در مغز موش¬های آلزایمری فعال نسبت به غیرفعال افزایش معناداری دارد. فعالیت ورزشی باعث افزایش سطح این شاخص های دفاعی می¬شود که این تغییرات با کاهش پروتئین¬های آپوپتوزی (سیتوکروم C، کسپیس-9، کسپیس-3 و بَکس) و افزایش پروتئین شوک گرمایی 70 (Heat Shock Protein 70) و عامل نروتروفیک مشتق از مغز (Brain Derived Neurotrophic Factor) مرتبط میباشد که با استفاده از فعالیت ورزشی منظم در مغز القاء شده و سپس با استفاده از کاهش بالینی پپتیدهای Aβ42 در موشهای آلزایمری، میانجیگری شدند (55). همچنین نشان داده شده است که 16 هفته دویدن روی نوارگردان به همراه مصرف اسید آلفالیپوئیک، سطوح Aβ42 مغز موشهای 13 ماهه آلزایمری تراریخته را کاهش داد. محققان بیان داشتند که افزایش استرس اکسایشی، یکی از عوامل اصلی درگیر در آلزایمر میباشد که افزایش تولید ROS را در پی دارد و باعث تخریب ساختارهای سلولی، افزایش تولید Aβ و در نهایت افزایش آپوپوتوز سلول میگردد. تمرین ورزشی به تنهایی و همراه با مصرف مکمل اسید آلفا لیپوئیک باعث افزایش سطح عوامل آنتیاکسیدانی به عنوان عوامل سرکوب کننده استرس اکسایشی شد و در نتیجه کاهش شاخصهای آپوپتوزی و Aβ را در پی دارد (54). همچنین تحقیق دیگری پیشنهاد میکند که فعالیت ورزشی منظم، آپوپتوز نورون عصبی را که در بیماریزایی آلزایمر دخالت دارند، از طریق افزایش تخریب یا پاکسازی Aβ، تضعیف میکند (56).
نتیجهگیری
در مجموع نتایج تحقیق حاضر نشان دهنده کاهش Aβ42 و MDA متعاقب تمرین تداومی و تناوبی در هیپوکامپ موشهای صحرایی نر سالمند میباشد. بنابراین احتمالاً تغییرات ایجاد شده در Aβ42 هیپوکامپ موشهای سالمند ممکن است در ارتباط با کاهش عوامل استرس اکسایشی ناشی از این تمرینات باشد و بنابراین تمرینات ورزشی تداومی و تناوبی میتواند از طریق تعدیل استرس اکسایشی و در نتیجه کاهش Aβ42، از تحلیل سیستم عصبی ناشی از سالمندی جلوگیری کند.
سپاسگزاری
این مقاله مستخرج از پایاننامه کارشناسی ارشد فیزیولوژی ورزش دانشگاه آزاد اسلامی واحد بجنورد میباشد. بدینوسیله محققین مراتب تشکر و قدردانی را از کلیه عزیزانی که در مراحل مختلف اجرای پژوهش همکاری و مساعدت داشتند، اعلام میدارند.
حامی مالی: ندارد.
تعارض در منافع: وجود ندارد.
همانند سایر سیستمهای بدن، تواناییهای عملکردی مغز در طی پیری به تدریج کاهش مییابد، که بهصورت کاهش در یادگیری و حافظه، توجه، سرعت تصمیمگیری، درک حسی (بینایی، شنوایی، لمس، بو و چشایی) و هماهنگی حرکتی آشکار میشود (1). یکی از مهمترین تغییرات ناشی از افزایش سن، کاهش عملکرد شناختی میباشد (2). روند زمانی کاهش عملکرد مغزی وابسته به سن، تقریباً به موازات کاهش عملکرد سایر سیستمهای بدن با شتاب قابلتوجهی، بعداز 50 سالگی، افزایش مییابد (3). بیماری آلزایمر بهعنوان شایعترین بیماری تحلیل برنده عصبی وابسته به سن میباشد؛ مطالعات جهانی از افزایش روز افزون این بیماری در میان جمعیت سالمند در جوامع مختلف دنیا نشان دارد و گفته میشود در حال حاضر 36 میلیون نفر مبتلا به آلزایمر در سراسر جهان هستند. پیشبینیها حاکی است که باتوجه به افزایش جمعیت سالمند در جهان، آمار مبتلایان به آلزایمر تا سال 2050 به 115 میلیون نفر خواهد رسید (4). یکی از عوامل اصلی ایجادکننده آلزایمر، تشکیل پلاکهای پیری متشکل از پپتید آمیلوئید بتا (Amyloid Beta) میباشد (5). آمیلوئید بتا بهصورت طبیعی در مقادیر اندک در مغز یافت میشود و دارای 49-37 اسید آمینه میباشد که در اثر پروتئولیز APP ایجاد میشود (6، 5). بیشترین آمیلوئید بتای ترشح شده، حاوی 40 اسید آمینه (Aβ40) است و درصد اندکی، حاوی 42 اسید آمینه (Aβ42) میباشد. آمیلوئید بتای 42، به دلیل حضور 2 اسید آمینه آب دوست بیشتر در ترکیبش، نسبت به Aβ40 آسانتر تجمع مییابد و سمیت بیشتری دارد (7). اشاره شده است که در یک مغز طبیعی Aβ40، حدود 90 درصد و Aβ42 تقریباً 10-5 درصد، پپتیدهای Aβ را تشکیل میدهند. پپتید Aβ42 اولین و اصلیترین پپتید برای تشکیل پلاکهای آمیلوئیدی میباشد. بنابراین اندازهگیری سطوح Aβ42 و نسبت Aβ42/Aβ40 به عنوان یک روش شناسایی افراد مستعد و مبتلا به آلزایمر مورد استفاده قرار میگیرد (8). علاوه بر این sojkova و همکاران (2011) در یک مطالعه طولی به مدت 1/5 سال نشان دادند که رسوب و پلاکهای Aβ مغز بهصورت وابسته به سن، در طی دوره تحقیق، حتی در سالمندان بدون علامت آلزایمر نیز افزایش مییابد (9). از طرفی نشان داده شده است که افزایش شاخصهای استرس اکسایشی در آلزایمر با تجمع و رسوب Aβ در مغز همراه است (10). آمیلوئید بتا در وضعیتهای تجمعی مختلفی مشاهده میشود که در این بین Aβ الیگومریزه شده بهعنوان سمی¬ترین شکل در نظر گرفته می-شود (11). Aβ الیگومریزه میتواند در میتوکندری نیز یافت شود (12) که مهمترین منبع تولید رادیکالهای آزاد میباشد. مطالعات در شرایط آزمایشگاهی نشان داده است که تزریق Aβ42 به نورونهای اولیه هیپوکامپی در محیط کاشت منجر به افزایش شاخصهای استرس اکسایشی و سمیت عصبی میشود (14،13). همراه با این استرس اکسایشی ناشی از پپتید Aβ، افزودن ویتامین E بهعنوان یک آنتی اکسیدان، بهطور معناداری استرس اکسایشی و اثرات سمیت عصبی ناشی از Aβ42 را تعدیل میکند (13) که این موضوع پیشنهاد میکند که سمیت عصبی ناشی از Aβ42 از طریق توانایی این پپتید سمی در ایجاد استرس اکسایشی، میانجیگری میشود. در این بین Casado و همکاران (2008) نشان دادند که سطح مالون دی آلدئید (Malondialdehyde) به عنوان شاخص پراکسیداسیون لیپیدی، همراه با افزایش سن در بیماران مبتلا به آلزایمر افزایش مییابد و استرس اکسایشی نقش مهمی در آسیبهای مغزی بیماران آلزایمری بازی میکند (15). شناسایی عواملی که سطح Aβ42 را در مغز تنظیم میکنند، هدف مهمی برای افزایش عملکرد و سلامت مغز میباشد (16). تمرین و فعالیت ورزشی یکی از راه کارهای حمایتی و غیرتهاجمی برای بهبود عملکرد مغز میباشد. یک مطالعه موردی نشان داده است که افراد مبتلا به اختلالات حافظه و یادگیری، در میانسالی کمتر فعال بودهاند و این غیرفعال بودن با 25 درصد افزایش توسعه آلزایمر همراه میباشد (17). به طور مشابه، تحقیق دیگری نشان داده است که فعالیت بدنی در میانسالی در برابر توسعه اختلالات شناختی، بیماری آلزایمر و جنون محافظت ایجاد میکند و فعالیت بدنی 60 درصد کاهش در شیوع آلزایمر در سالمندی را به همراه دارد (18). Yu و همکاران (2013) نشان دادند که تمرین ورزشی از طریق کاهش معنادار در سطح Aβ42، کاهش سطح MDA و بهبود فعالیت سوپراکساید دیسموتاز (Superoxide dismutase)، را در پی داشته است که با بهبود حافظه و سلامت مغز همراه است (19). Zhao و همکاران (2015) نشان دادند که 5 ماه تمرین ورزشی نوارگردان از تجمع Aβ (در سن 17 ماهگی) جلوگیری میکند و بنابراین میتوان از تمرین نوارگردان برای کند کردن پیشرفت آلزایمر بعد از مرحله تجمع پلاکهای آمیلوئیدی استفاده کرد (20). همچنین یعقوبی و همکاران (1395) عنوان داشتند که تمرین ورزشی تداومی باعث کاهش Aβ42 هیپوکامپ موشهای آلزایمری میشود (21). تحقیقات مختلفی به بررسی سازوکارهای درگیر در تعدیل سطح Aβ42 متعاقب فعالیت ورزشی با شدت پایین و متوسط را مورد بررسی قرار دادهاند (23، 22) ولی اثر پروتکل های تمرین طولانیمدت و سازوکارهای احتمالی ناشی از آن به خوبی درک نشده است. در نتیجه نتایج تحقیق حاضر برای یافتن استراتژیهای بالقوه و طراحی تمرین و یافتن پروتکل تمرین مناسب برای پیشگیری از انحطاط عصبی ناشی از سالمندی مفید خواهد بود. بنابراین هدف از تحقیق حاضر بررسی اثر تمرینات تداومی و تناوبی بر سطح Aβ42 و MDA در هیپوکامپ موشهای صحرایی نر سالمند بود.
روش بررسی
پژوهش حاضر از نوع تجربی با طرح پس آزمون به همراه گروه کنترل بود که به شیوه آزمایشگاهی انجام شد. در این تحقیق از 30 سر موش سالمندنر نژاد ویستار با دامنه وزنی 320 تا 380 گرم و سن 18 ماهه (24) استفاده شد که از آزمایشگاه حیوانی دانشگاه آزاد اسلامی واحد مرودشت خریداری گردید. مطابق با خطمشی انجمن ایرانیان حمایت از حیوانات آزمایشگاهی در قفسهای 4تایی و تحت شرایط استاندارد (چرخه 12 ساعته روشنایی- تاریکی، دمای 2±25 درجه سانتیگراد) با دسترسی آزاد به آب و غذا نگهداری شدند. پس از یک هفته آشنایی و سازگاری با محیط جدید، موشهای صحرایی نر سالمند بهطور تصادفی و بر اساس وزن به سه گروه تمرین تناوبی، تمرین تداومی و کنترل با تعداد برابر در هر گروه (10 سر) تقسیم شدند. در ادامه، موشهای نر سالمند با راه رفتن و دویدن بر روی نوارگردان آشنا شدند. پس از یک هفته آشناسازی، حداکثر سرعت دویدن با استفاده از آزمون عملکرد ورزشی مدرج برآورد شد. آنگاه، موشهای گروههای تجربی در هشت هفته تمرینات تناوبی یا تمرین تداومی دویدن روی نوارگردان شرکت کردند. 48 ساعت پس از آخرین جلسه تمرینی و پس از ناشتایی شبانه، تمامی موشهای بیهوش شده و بافتبرداری صورت گرفت. لازم به ذکر است تمامی موشهای در طول مداخله رژیم غذایی استاندارد را مصرف کردند. تمامی مداخلات حیوانی مطابق با دستورالعملهای اخلاقی مؤسسات ملی برای مراقبت و استفاده از حیوانات آزمایشگاهی انجام شد. پروتکل تمرین تداومی به مدت هشت هفته و 5 جلسه در هفته بر روی نوارگردان (شیب صفر درجه) اجرا شد. پروتکل تمرین تداومی در 3 مرحله آشنایی، اضافه بار و تثبیت بار اجرا شد. در مرحله آشنایی (هفته اول)، موشها هر روز به مدت 15-10 دقیقه با سرعت 10 متر در دقیقه روی نوارگردان راه رفتند. در مرحله اضافه بار (هفته دوم تا پنجم)، به تدریج در طی 3 هفته، به شدت و مدت فعالیت افزوده شد؛ تا به میزان نهایی 40 دقیقه با سرعت 20 متر در دقیقه رسید. در مرحله حفظ یا تثبیت (هفته ششم تا هشتم)، تمرین با همین شدت ادامه یافت تا 8 هفته به پایان رسید. این پروتکل با شدت 65 تا 70% VO2max موشهای سالمند بود. همچنین، 10 دقیقه گرمکردن و 5 دقیقه سردکردن با شدت پایین در ابتدا و انتهای هر جلسه تمرینی اجرا شد (25). پروتکل تمرین تناوبی نیز به مدت هشت هفته و 5 جلسه در هفته بر روی نوارگردان (شیب صفر درجه) اجرا شد. این پروتکل، شامل اجرای 8-5 مرحله فعالیت 2 دقیقهای با شدت معادل 80 تا 100 درصد اکسیژن مصرفی بیشینه و با دورههای استراحتی فعال 2 دقیقهای با شدت معادل 50 درصد اکسیژن مصرفی بیشینه بود. بر این اساس، سرعت دویدن در هفته اول از 22 متر بر دقیقه به 31 متر بر دقیقه در دو هفته پایانی رسید. همچنین، تعداد تکرار تناوب در هفته اول از 5 تکرار به 8 تکرار در هفته چهارم رسید و در ادامه نیز با 8 تکرار اجرا شد. همچنین، 10 دقیقه گرمکردن و 5 دقیقه سردکردن با شدت پایین در قبل و بعد از هر جلسه تمرینی در نظر گرفته شد (27-25). تمامی آزمودنیها، 72 ساعت پس از آخرین جلسه تمرین، با تزریق درون صفاقی ترکیبی از کتامین (50 میلیگرم به ازای هر کیلوگرم) و زایلازین (4 میلیگرم به ازای هر کیلوگرم) بیهوش شدند. برای جمعآوری نمونههای هیپوکامپ، سر آزمودنی ها از ناحیه گردن توسط قیچی مخصوص جدا شد، ابتدا با استفاده از تیغ جراحی، جمجمه شکافته شده و مغز با احتیاط خارج گردید. مغز سالم توسط تیغ جراحی دقیقاً از وسط به دو نیم تقسیم شد و با توجه به مختصات هیپوکامپ به کمک اطلس پاک سینوس، هیپوکامپ از سیستم لمبیک جدا شد. نمونههای هیپوکامپ جمعآوری شده برای اندازهگیریهای بعدی در دمای ۸۰- درجه سانتیگراد نگهداری شد. برای اندازهگیری مقادیر پروتئینی Aβ42 و MDA از روش وسترن بلات استفاده شد. برای استخراج پروتئین های هیپوکامپ از بافر سنجش رسوب رادیوایمنی (Radioimmunoprecipitation Assay Buffer) حاوی 0/05 میلیمولار بافر تریس ( pHبرابر 8)، 150 میلیمولار کلرید سدیم، 0/01 درصد اتیلن گلیکول تترااسید استیک (Ethylene Glycol Tetra Acetic Acid))، یک درصد SDSبه اضافه 0/1 درصد آنتی پروتئاز کوکتیل (Protease Inhibitor Cocktail) استفاده شد. به این ترتیب که 100 میلیگرم بافت در 500 میکرولیتر بافر حاوی آنتی پروتئاز توسط یک هموژنایزر دستی هموژن شد و نیم ساعت در دمای 4 درجه سانتیگراد گذاشته شدند و سپس دریک سانتریفوژ یخچال دار (bo ,sw14rfroil) در دور 12000 و 4 درجه سانتیگراد و به مدت 10 دقیقه سانتریفوژ شد. مایع رویی جمعآوری شده و غلظت پروتئین آن با کیت تعیینکننده پروتئین (Bio-Rad) در طول موج 595 نانومتر اندازهگیری و در نهایت در دمای 20 درجه زیر صفر در فریزر نگهداری شد. سپس، هموژن به دست آمده به نسبت 1:1 با نمونه لودینگ بافر (mM 50تریس-کلرید هیدروژن، 2 درصد سدیم دو دسیل سولفات، 10 درصد گلیسرول، 5 درصد بتا- مرکاپتواتانول و 0/005 درصد برموفنول آبی) مخلوط گردید. آنگاه، نمونهها به مدت 5 دقیقه جوشانده شد تا تمام پروتئینها کاملاً دناتوره شوند. پروتئینها با استفاده از الکتروفورز ژل SDS-polyacrylamide جدا شده و به غشای نیتروسلولز منتقل شدند. غشا به مدت 1 ساعت در 5 درصد BSA در Tris-Buffered Saline و 0/1 درصد Tween 20 TBST)) مسدود شد و در آنتیبادی اولیه (1: 500) انکوبه شد. انکوباسیون در آنتیبادی ثانویه روز بعد به مدت 1 ساعت در دمای اتاق در 4 درصد TBSTانجام شد. پروتئینها با یک واکنش شیمیایی لومینسانس و با تجزیه و تحلیل densitometry با نرمافزار Image J اندازهگیری شدند. آنتیبادیهای اولیه و ثانویه Aβ42 (F-4):sc-390904 و mouse anti-rabbit IgG-HRP:sc-2357 از شرکتSanta Cruz Biotechnology, Inc مورد استفاده قرار گرفتند. سطح MDA به عنوان شاخصی از پراکسیداسیون لیپیدی در هیپوکامپ موشهای سالمند با روش جذب سنجی با استفاده از کیت شرکت زلبایو ساخت کشور آلمان در دامنه 532 (540-530) نانومتر اندازهگیری شد. بر اساس مقایسه با نمودار استاندارد نتایج حاصل، میزان MDA بر حسب نانو مول بر میلیگرم پروتئین بیان شد.
تجزیه و تحلیل آماری
از آزمون شاپیروولیک جهت بررسی توزیع طبیعی دادهها استفاده شد. جهت تعیین معنادار بودن تفاوت میانگین پس آزمون متغیرها بین گروههای تحقیق، از آزمون آنالیز واریانس یک طرفه استفاده گردید. اطلاعات مورد نیاز پس از جمعآوری، توسط نرمافزار آماری version 16 SPSS در سطح معناداری p< 0/05 مورد تجزیه و تحلیل قرار گرفت.
ملاحظات اخلاقی
پروپوزال این تحقیق توسط دانشگاه آزاد اسلامی واحد بجنورد تایید شده است (کد اخلاق IR.IAU.BOJNOURD.REC.1399.022).
نتایج
در این بخش اطلاعات توصیفی مربوط به وزن موشها در ابتدا و انتهای دوره پژوهش ارائه شده است. جهت اطمینان از همگن بودن گروهها از نظر وزن در پیش آزمون از آزمون آنالیز واریانس یک طرفه استفاده شد که نتایج آن در یک ستون در جدول 1 قرار داده شده است. تعداد نمونهها در تمام گروهها 10 سر بود که در طی دوره پژوهش با ریزش در گروه تمرین تناوبی و کنترل همراه بود که در نهایت در هر گروه 9 سر باقی ماندند ولی در موشهای تعداد گروه تمرین تداومی تغییری ایجاد نشد. با عنایت به جدول فوق و سطح معناداری گزارش شده میتوان دریافت گروهها در گروه بندی اولیه و انتهای دوره تحقیق تفاوت معناداری با یکدیگر نداشتند (0/05<p). به منظور مقایسه سطوح پروتئینی Aβ42 و MDA هیپوکامپ در گروههای تحقیق از آزمون آنووا (ANOVA) و آزمون تعقیبی توکی استفاده شد. در جدول 2 یافتههای آزمون آماری در خصوص مقایسه اثر تمرین تمرین تداومی و تمرین تناوبی بر سطح Aβ42 و MDA هیپوکامپ موشهای صحرایینر سالمند در گروههای تحقیق ارائه شده است. سطوح پروتئینی Aβ42 هیپوکامپ گروههای تحقیق. بر اساس نتایج حاصل از آزمون تعقیبی توکی، سطح پروتئین Aβ42 هیپوکامپ در گروه تمرین تداومی بهطور معناداری نسبت به گروه کنترل پایینتر بود (0/001=p). همچنین سطح این شاخص در گروه تمرین تناوبی بهطور معناداری نسبت به گروه کنترل پایینتر بود (0/001=p). ولی بین سطح پروتئین Aβ42 هیپوکامپ در گروه تمرین تداومی و تمرین تناوبی تفاوت معناداری مشاهده نشد (0/502=p). سطوح پروتئینی Aβ42 هیپوکامپ گروه¬های تحقیق، در نمودار 1 ارائه شده است. سطوح MDA هیپوکامپ گروههای تحقیق. بر اساس نتایج حاصل از آزمون تعقیبی توکی، سطح MDAهیپوکامپ در گروه تمرین تداومی بهطور معناداری نسبت به گروه کنترل پایینتر بود (0/016=p). همچنین سطح این شاخص در گروه تمرین تناوبی بهطور معناداری نسبت به گروه کنترل پایینتر بود (0/046=p). ولی بین سطح MDA هیپوکامپ در گروه تمرین تداومی و تمرین تناوبی تفاوت معناداری مشاهده نشد (0/866=p). سطوح MDA هیپوکامپ گروه¬های تحقیق، در نمودار 2 ارائه شده است.
جدول 1: مقایسه وزن آزمودنیها در پیش آزمون و پس از 8 هفته تمرین تداومی و تناوبی در گروههای تحقیق
* جهت مقایسه گروه ها از آزمون آنالیز واریانس یک طرفه استفاده شد.
جدول 2: مقایسه سطوح Aβ42 و MDA در هیپوکامپ گروههای تحقیق و یافتههای آزمون آنالیز واریانس
* نشانه تفاوت معنادار بین گروههای تحقیق در سطح 0/05>p.
نمودار1: مقایسه سطوح پروتئینی Aβ42 هیپوکامپ گروههای تحقیق پس از 8 هفته تمرین تناوبی و تداومی.
*نشانه تفاوت معنی دار نسبت به گروه کنترل در سطح 0/05>p
نمودار 2: مقایسه سطوح MDA هیپوکامپ گروههای تحقیق پس از 8 هفته تمرین تناوبی و تداومی.
*نشانه تفاوت معنی دار نسبت به گروه کنترل در سطح 0/05>p
بحث
نتایج تحقیق حاضر نشان داد که تمرین ورزشی فارغ از شدت و نوع تمرین سطح Aβ42 و MDA هیپوکامپ موشهای سالمند را کاهش داد. دو عامل اصلی بیماری آلزایمر شامل تشکیل پلاکهای پیری متشکل از پپتید Aβ و کلافههای نوروفیبریلار (Neurofibrillary tangle) متشکل از پروتئینهای هیپرفسفوریله تائو (Tau hyperphosphorylated) میباشد (5). ماهیت آب گریزی پپتیدهای Aβ42، به آنها اجازه میدهدکه تجمع یافته و پروتوفیبریلها و فیبریلها را تشکیل دهند و سرانجام تجمع این عوامل منجر به رسوب پلاک¬های آمیلوئیدی را می¬دهند (28). افزایش تولید Aβ42 و کاهش حذف آن از مغز عامل اصلی تجمع آن در مغز و آسیب به ارتباطات نرونی و کاهش حافظه و یادگیری بیان شده است (29). در این ارتباط بیان شده است که افزایش سطح و رسوب Aβ در پلاکهای خارج سلول بهطور ویژه باعث اختلال سیناپسی، اختلال در شبکه نورونی، اختلال در عملکرد میتوکندریایی، آپوپتوز سلول عصبی و کاهش حافظه میگردد (32-30). همچنین، هر دوی میکروگلیاها و آستروسیتها، پروتئین Aβ ترشح میکنند (30). پلاکهای پیری اصولاً متشکل از پپتیدهای Aβ می¬باشد که نقش اصلی آن در بیماریزایی آلزایمر به طور گستردهای ثابت شده است (33، 30). در حال حاضر شواهد وجود دارند مبنی بر اینکه تمرینات ورزشی، یک عامل مهم در برابر روند سالمندی و بیماریهای تحلیل برنده عصبی ناشی از آن میباشد (34). در مطالعه حاضر سطح Aβ42 هیپوکامپ موشها در هر دو تمرین تداومی و تناوبی نسبت به موش¬های گروه کنترل، بهطور معناداری پایینتر بود. نتایج مربوط به تأثیر تمرین تداومی و تناوبی نشان داد که سطح Aβ42 تفاوت معناداری بین دو پروتکل تمرینی وجود ندارد. این نتایج با نتایج Kang & Cho (2014) که نشان دادند 6 هفته تمرین نوارگردان از طریق بهبود سیگنال دهی انسولین، کاهش معنادار سطح Aβ42 را در مغز موش¬های آلزایمری را باعث شده است (35). همچنینLiu و همکاران (2013) نشان دادند که تمرین میتواند از طریق اثر مهاری بر سطح Aβ، به عنوان راهکار درمانی برای آلزایمر مطرح باشد (36). همچنین Kang و همکاران (2013) بیان داشتند که 12 هفته تمرین ورزشی روی نوارگردان از اختلال جهش در ژن PS2 جلوگیری کرده و تجمع Aβرا از طریق مهار فعالیت β-سکرتاز و فراورده های آن، کاهش داد (37). همچنین یعقوبی و همکاران (1394) با بررسی تأثیر تمرین تداومی با دو شدت پایین و بالا بر موشهای صحرایی آلزایمری شده عنوان داشتند که تمرین تداومی فارغ از شدت تمرین (پایین و بالا) باعث کاهش سطح Aβ42 هیپوکامپ موشهای آلزایمری شده میگردد (21). درباره تغییرات سطح Aβو متابولیسم آن در اثر تمرین، نظریههای مختلفی وجود دارد. با توجه به این که فعالیت ورزشی بسیاری از فرآوردههای ژنی را هم در سطح mRNA و هم در سطح پروتئین تعدیل می¬کند، القاءکننده تغییرات آناتومیکی، عصبیشیمیایی و الکتروفیزیولوژیکی افزایش دهنده شکلپذیری نورونی میباشد (38)، این احتمال وجود دارد که چندین مسیر برای تنظیم سطح Aβ بهطور مستقیم و یا به طور غیرمستقیم فعال باشد. به عنوان مثال یک روش احتمالی، افزایش فعالیت پروتئازوم (Proteasome) ناشی از ورزش است که قبلاً در عضله اسکلتی گزارش شده است (39) و می¬تواند تخریب قطعات پروتئولیتیکی APP را میانجیگریکند (40). اما Adlard و همکاران (2005) بیان داشتند که احتمالاً تمرین ورزشی می-تواند متابولیسم APP و آبشار Aβ در مغز را در راستای کاهش تولید Aβ میانجیگری کند که مستقل از سطح نپریلیزین و IDE به عنوان عوامل اصلی تجزیه Aβ42، میباشد (41). احتمال دوم این است که ورزش به طور مستقیم متابولیسم APP را با استفاده از افزایش فعالیت نورونی تعدیل میکند. به عنوان مثال، پردازش APP میتواند توسط پروتئینکیناز فعال¬شده با میتوژن (Mitogen-Activated Protein Kinases) و فسفولیپاز C تکمیل شود و ثابت شده است که این مسیرها از طریق ورزش فعال میشوند (42). از طرف دیگر، فعالیت کولینرژیک با ورزش افزایش مییابد و تنظیم سیستم کولینرژیک توسط ورزش در شکل¬پذیری نورونی ناشی از ورزش دخالت دارد (38). نقش MDA به عنوان یک شاخص از آسیب اکسایشی ناشی از بیماری و سالمندی به خوبی ثابت شده است (43). در مطالعه Gil و همکاران (2006) که 194 زن و مرد سالم بین سنین 18 تا 84 سال مورد بررسی قرار گرفتند، نشان داده شد که سطح MDA پلاسما با افزایش سن افزایش مییابد که نشان دهنده تسریع اکسیداسیون در طی سالمندی میباشد (44). نتایج محققان هندی که به بررسی 100 مرد سالم پرداختند نیز نشان میدهد که نسبت MDA/TAC (total antioxidant capacity) با افزایش سن افزایش می یابد (45). همچنین Dei و همکاران (2002) نشان دادند که سطح MDA در اطراف آستروسیتها، نورون ها و پلاکهای هیپوکامپی بیماران آلزایمری افزایش مییابد (46). دبیدی روشن و همکاران (2013)، در ارتباط با تأثیر تمرین ورزشی استقامتی بر استرس اکسایشی، بیان داشتند 8 هفته تمرین استقامتی باعث کاهش MDA به عنوان شاخص پراکسیداسیون لیپیدی در هیپوکامپ موشهای آلوده به سرب میشود (47). همچنین فلاح محمدی و همکاران (1390) نشان دادند که به دنبال اجرای هشت هفته تمرینات تداومی آثار تخریبی القائی توسط هموسیستئین در هیپوکامپ موشهای صحرایی کاهش یافته است که بهوسیله کاهش سطوح MDA و افزایش سطوح SOD مشخص شد (48). در زمینه ارتباط استرس اکسایشی و آسیبهای سیستم عصبی نیز ارتباط نزدیکی بین MDA و Aβ42 مورد بررسی قرار گرفته است و بهطور گستردهای ثابت شده است که تجمع پپتید Aβ در مغز باعث القای استرس اکسایشی و التهاب عصبی می¬گردد (33،30). شواهدی وجود دارد که Aβ42 داخل نورونی میتواند وارد میتوکندری شده، زنجیره انتقال الکترونی را مختل و تولید گونههای اکسیژن واکنش دهنده شامل رادیکالهای هیدروکسیل، سوپراکسید و پراکسیداسیون هیدروژن را القا کند (49). همچنین مطالعات در شرایط آزمایشگاهی نشان داده است که تزریق Aβ42 به نورونهای اولیه هیپوکامپی در محیط کاشت منجر به افزایش شاخصهای استرس اکسایشی و سمیت عصبی میشود (14،13). همراه با این استرس اکسایشی ناشی از پپتید Aβ، افزودن ویتامین E به عنوان یک آنتیاکسیدان، بهطور معناداری استرس اکسایشی و اثرات سمیت عصبی ناشی از Aβ42 را تعدیل میکند (13)، این موضوع پیشنهاد می¬کند که سمیت عصبی ناشی از Aβ42 از طریق توانایی این پپتید سمی در ایجاد استرس اکسایشی، میانجیگری میشود و بالعکس افزایش استرس اکسایشی و تخلیه آنتیاکسیدانی، افزایش سطح این شاخص را در مغز افراد آلزایمری در پی دارد. در این راستا تحقیق دیگری نشان داد که تخلیه ویتامین E، تجمع Aβ را از طریق کاهش پاکسازی از مغز و خون در موش¬های آلزایمری، افزایش میدهد (14). همچنین نشان داده شده است که تزریق Aβ40 به درون مغز موشها، با القای آسیب رادیکال آزاد و تغییرات در دفاع آنتیاکسیدانی مثل تخلیه گلوتاتیون در قشر پیشپیشانی و هیپوکامپ موش¬ها ارتباط دارد (50). علاوه بر این نشان داده شده است که مصرف مکملهای آنتی اکسیدانی و یا کاهش استرس اکسایشی کاهش سطح Aβ42 و بهبود عملکرد شناختی را در پی دارد. در این ارتباط 5 ماه خوراندن اسید گالیک و عصاره پلی¬فنولی دانه انگور غنی از کاتچین به موش-های آلزایمری، از بدتر شدن شناخت جلوگیری کرده و موجب کاهش سطوح اُلیگومرهای Aβ می¬شود (51). مکانیسم احتمالی موجود در این باب عبارت است از اینکه کاهش استرس اکسایشی و افزایش دفاع آنتیاکسیدانی به عنوان عامل افزایش دهنده فعالیت آنزیم α-سکرتاز و بازدارنده β- و γ-سکرتاز عمل می-کنند و بنابراین پردازش APPرا به سمت مسیر غیرآمیلوئیدوژنیک هدایت میکنند (52). از طرف دیگر بیان شده است که فعالیت ورزشی منظم ظرفیت آنتیاکسیدانی را بهبود می¬بخشد که در نتیجه منجر به جلوگیری از استرس اکسایشی، آپوپتوز و آسیب سلولی می¬شود و در مجموع احتمال خطر زوال عقل یا آلزایمر را کاهش میدهد (54، 53). در این راستا Um و همکاران (2008) نشان دادند که سطح SOD-1 و پروتئین کاتالاز در مغز موش¬های آلزایمری فعال نسبت به غیرفعال افزایش معناداری دارد. فعالیت ورزشی باعث افزایش سطح این شاخص های دفاعی می¬شود که این تغییرات با کاهش پروتئین¬های آپوپتوزی (سیتوکروم C، کسپیس-9، کسپیس-3 و بَکس) و افزایش پروتئین شوک گرمایی 70 (Heat Shock Protein 70) و عامل نروتروفیک مشتق از مغز (Brain Derived Neurotrophic Factor) مرتبط میباشد که با استفاده از فعالیت ورزشی منظم در مغز القاء شده و سپس با استفاده از کاهش بالینی پپتیدهای Aβ42 در موشهای آلزایمری، میانجیگری شدند (55). همچنین نشان داده شده است که 16 هفته دویدن روی نوارگردان به همراه مصرف اسید آلفالیپوئیک، سطوح Aβ42 مغز موشهای 13 ماهه آلزایمری تراریخته را کاهش داد. محققان بیان داشتند که افزایش استرس اکسایشی، یکی از عوامل اصلی درگیر در آلزایمر میباشد که افزایش تولید ROS را در پی دارد و باعث تخریب ساختارهای سلولی، افزایش تولید Aβ و در نهایت افزایش آپوپوتوز سلول میگردد. تمرین ورزشی به تنهایی و همراه با مصرف مکمل اسید آلفا لیپوئیک باعث افزایش سطح عوامل آنتیاکسیدانی به عنوان عوامل سرکوب کننده استرس اکسایشی شد و در نتیجه کاهش شاخصهای آپوپتوزی و Aβ را در پی دارد (54). همچنین تحقیق دیگری پیشنهاد میکند که فعالیت ورزشی منظم، آپوپتوز نورون عصبی را که در بیماریزایی آلزایمر دخالت دارند، از طریق افزایش تخریب یا پاکسازی Aβ، تضعیف میکند (56).
نتیجهگیری
در مجموع نتایج تحقیق حاضر نشان دهنده کاهش Aβ42 و MDA متعاقب تمرین تداومی و تناوبی در هیپوکامپ موشهای صحرایی نر سالمند میباشد. بنابراین احتمالاً تغییرات ایجاد شده در Aβ42 هیپوکامپ موشهای سالمند ممکن است در ارتباط با کاهش عوامل استرس اکسایشی ناشی از این تمرینات باشد و بنابراین تمرینات ورزشی تداومی و تناوبی میتواند از طریق تعدیل استرس اکسایشی و در نتیجه کاهش Aβ42، از تحلیل سیستم عصبی ناشی از سالمندی جلوگیری کند.
سپاسگزاری
این مقاله مستخرج از پایاننامه کارشناسی ارشد فیزیولوژی ورزش دانشگاه آزاد اسلامی واحد بجنورد میباشد. بدینوسیله محققین مراتب تشکر و قدردانی را از کلیه عزیزانی که در مراحل مختلف اجرای پژوهش همکاری و مساعدت داشتند، اعلام میدارند.
حامی مالی: ندارد.
تعارض در منافع: وجود ندارد.
References:
1- Levin O, Fujiyama H, Boisgontier MP, Swinnen SP, Summers J. Aging and Motor Inhibition: A Converging Perspective Provided by Brain Stimulation and Imaging Approaches. Neurosci Biobehav Rev 2014; 43: 100-17.
2- Alexander GE, Ryan L, Bowers D, Foster TC, Bizon JL, Geldmacher DS, et al. Characterizing Cognitive Aging in Humans with Links to Animal Models. Front Aging Neurosci 2012; 4: 21.
3- Mendonca GV, Pezarat-Correia P, Vaz JR, Silva L, Heffernan KS. Impact of Aging on Endurance and Neuromuscular Physical Performance: The Role of Vascular Senescence. Sports Med 2017; 47(4): 583-98.
4- Khaledi S, Ahmadi S. Amyloid Beta and Tau: From Physiology to Pathology in Alzheimer’s Disease. Neurosci J Shefaye Khatam 2016; 4(4): 67-88 .[Persian]
5- Selkoe DJ. Alzheimer's Disease: Genotypes, Phenotype, and Treatments. Science 1997; 275(5300): 630.
6- Gispen WH, Biessels GJ. Cognition and Synaptic Plasticity in Diabetes Mellitus. Trends Neurosci 2000; 23(11): 542-9.
7- Zafra F, Lindholm D, Castren E, Hartikka J, Thoenen H. Regulation of Brain-Derived Neurotrophic Factor and Nerve Growth Factor Mrna in Primary Cultures of Hippocampal Neurons and Astrocytes. J Neurosci 1992; 12(12): 4793-9.
8- Dries DR, Yu G. Assembly, Maturation, And Trafficking of the γ-Secretase Complex in Alzheimer’s Disease. Curr Alzheimer Res 2008; 5(2): 132-46.
9- Sojkova J, Zhou Y, An Y, Kraut MA, Ferrucci L, Wong DF, et al. Longitudinal Patterns of β-Amyloid Deposition in Nondemented Older Adults. Arch Neurol 2011; 68(5): 644-9.
10- Butterfield DA, Sultana R. Methionine-35 of Aβ (1–42): Importance for Oxidative Stress in Alzheimer Disease. J Amino Acids 2011; 2011: 198430.
11- Glabe CC. Amyloid Accumulation and Pathogensis of Alzheimer’s Disease: Significance of Monomeric, Oligomeric and Fibrillar Aβ. Subcell Biochem 2005; 38: 167-77.
12- Caspersen C, Wang N, Yao J, Sosunov A, Chen X, Lustbader JW, et al. Mitochondrial Abeta: A Potential Focal Point for Neuronal Metabolic Dysfunction in Alzheimer's Disease. FASEB J 2005; 19(14): 2040-1.
13- Boyd-Kimball D, Mohmmad Abdul H, Reed T, Sultana R, Butterfield DA. Role of Phenylalanine 20 Inalzheimer's Amyloid β-Peptide (1-42)-Induced Oxidative Stress and Neurotoxicity. Chem Res Toxicol 2004; 17(12): 1743-9.
14- Nishida Y, Ito S, Ohtsuki S, Yamamoto N, Takahashi T, Iwata N, et al. Depletion of Vitamin E Increases Amyloid β Accumulation by Decreasing its Clearances from Brain and Blood in a Mouse Model of Alzheimer Disease. J Biol Chem 2009; 284(48): 33400-8.
15- Casado A, López-Fernández ME, Casado MC, de La Torre R. Lipid Peroxidation and Antioxidant Enzymeactivities in Vascular and Alzheimer Dementias. Neurochem Res 2008; 33(3): 450-8.
16- Berchtold N, Chinn G, Chou M, Kesslak J, Cotman C. Exercise Primes a Molecular Memory for Brain-Derived Neurotrophic Factor Protein Induction in the Rat Hippocampus. Neurosci 2005; 133(3): 853-61.
17- Friedland RP, Fritsch T, Smyth KA, Koss E, Lerner AJ, Chen CH, et al. Patients with Alzheimer's Disease Have Reduced Activities in Midlife Compared with Healthy Control-Group Members. Proc Natl Acad Sci Usa 2001; 98(6): 3440-5.
18- Laurin D, Verreault R, Lindsay J, Macpherson K, Rockwood K. Physical Activity and Risk of Cognitive Impairment and Dementia in Elderly Persons. Arch Neurol 2001; 58(3): 498-504.
19- Yu F, Xu B, Song C, Ji L, Zhang X. Treadmill Exercise Slows Cognitive Deficits in Aging Rats by Antioxidation and Inhibition of Amyloid Production. Neurorep 2013; 24(6): 342-7.
20- Zhao G, Liu H, Zhang H, Tong X. Treadmill Exercise Enhances Synaptic Plasticity, But Does Not Alter β-Amyloid Deposition in Hippocampi of Aged APP/PS1 Transgenic Mice. Neurosci 2015; 298: 357-66.
21- Yaghoubi A, Saghebjoo M, Fallah Mohammadi Z, Hedayati M, Hajizadeh-Moghaddam A. Effects of Continuous Training Intensity on Amyloid Beta1-42 (Aβ1-42) Levels in Hippocampus of Homocysteine-Induced Alzheimer's Model Rats. J Arak Uni Med Sci 2016; 18(11): 83-93.[Persian]
22- Seifert T, Brassard P, Wissenberg M, Rasmussen P, Nordby P, Stallknecht B, et al. Endurance Training Enhances BDNF Release from the Human Brain. Am J Physiol Regul Integr Comp Physiol 2010; 298(2): R372-7.
23- Groover AL, Ryals JM, Guilford BL, Wilson NM, Christianson JA, Wright DE. Exercise-Mediated Improvements in Painful Neuropathy Associated with Prediabetes in Mice. Pain 2013; 154(12): 2658-67.
24- Ziaaldini MM, Koltai E, Csende Z, Goto S, Boldogh I, Taylor AW, et al. Exercise Training Increases Anabolic and Attenuates Catabolic and Apoptotic Processes in Aged Skeletal Muscle of Male Rats. Exp Gerontol 2015; 67: 9-14.
25- Hafstad AD, Lund J, Hadler-Olsen E, Höper AC, Larsen TS, Aasum E. High- and Moderate-Intensity Training Normalizes Ventricular Function and Mechanoenergetics in Mice with Diet-Inducedobesity. Diabetes 2013; 62(7): 2287-94.
26- Hafstad AD, Boardman NT, Lund J, Hagve M, Khalid AM, Wisløff U, et al. High Intensity Interval Training Alters Substrate Utilization and Reduces Oxygen Consumption in The Heart. J Appl Physiol 2011; 111(5): 1234-41.
27- Azali Alamdari K, Khalafi M. The Effects of High Intensity Interval Training on Serum Levels of FGF21 and Insulin Resistance in Obese Men. IJDM 2019; 18(1): 41-8.[Persian]
28- Viola KL, Klein WL. Amyloid Βoligomers in Alzheimer’s Disease Pathogenesis, Treatment, and Diagnosis. Acta Neuropathol 2015; 129(2): 183-206.
29- Selkoe DJ, Hardy J. The Amyloid Hypothesis of Alzheimer's Disease at 25 Years. EMBO Mol Med 2016; 8(6): 595-608.
30- Uslu S, Akarkarasu ZE, Ozbabalik D, Ozkan S, Çolak O, Demirkan ES, et al. Levels of Amyloid Beta-42, Interleukin-6 and Tumor Necrosis Factor-Alpha in Alzheimer’s Disease and Vascular Dementia. Neurochem Res 2012; 37(7): 1554-9.
31- Capetillo-Zarate E, Gracia L, Tampellini D, Gouras GK. Intraneuronal Aβ Accumulation, Amyloid Plaques, And Synapse Pathology in Alzheimer’s Disease. Neurodegener Dis 2012; 10(1-4): 56-9.
32- Cavallucci V, D’amelio M, Cecconi F. Aβ Toxicity in Alzheimer's Disease. Mol Neurobiol 2012; 45(2): 366-78.
33- Souza LC, Carlos Filho B, Goes AT, Del Fabbro L, de Gomes MG, Savegnago L, et al. Neuroprotective Effect of Physical Exercise in a Mouse Model of Alzheimer’s Disease Induced by β-Amyloid1–40 Peptide. Neurotox Res 2013; 24(2): 148-63.
34- Paillard T, Rolland Y, de Souto Barreto P. Protective Effects of Physical Exercise in Alzheimer's Disease and Parkinson's Disease: A Narrative Review. J Clin Neurol 2015; 11(3): 212-9.
35- Kang EB, Cho JY. Effects of Treadmill Exercise on Brain Insulin Signaling and β-Amyloid in Intracerebroventricular Streptozotocin Induced-Memory Impairment in Rats. J Exerc Nutrition Biochem 2014; 18(1): 89-96.
36- Liu H-L, Zhao G, Zhang H, Shi LD. Long-Term Treadmill Exercise Inhibits the Progression of Alzheimer's Disease-Like Neuropathology in the Hippocampus of APP/PS1 Transgenic Mice. Behav Brain Res 2013; 256: 261-72.
37- Kang EB, Kwon IS, Koo JH, Kim EJ, Kim CH, Lee J, et al. Treadmill Exercise Represses Neuronal Cell Death and Inflammation During Aβ-Induced ER Stress by Regulating Unfolded Protein Response in Aged Presenilin 2 Mutant Mice. Apoptosis 2013; 18(11): 1332-47.
38- Cotman CW, Berchtold NC. Exercise: A Behavioral Intervention to Enhance Brain Health and Plasticity. Trends Nurosci 2002; 25(6): 295-301.
39- Radák Z, Sasvári M, Nyakas C, Taylor AW, Ohno H, Nakamoto H, et al. Regular Training Modulates the Accumulation of Reactive Carbonyl Derivatives in Mitochondrial and Cytosolic Fractions of Rat Skeletal Muscle. Arch Biochem Biophys 2000; 383(1): 114-8.
40- Schmitz A, Schneider A, Kummer MP, Herzog V. Endoplasmic Reticulum‐Localized Amyloid β‐Peptide is Degraded in the Cytosol by Two Distinct Degradation Pathways. Traffic 2004; 5(2): 89-101.
41- Adlard PA, Perreau VM, Pop V, Cotman CW. Voluntary Exercise Decreases Amyloid Load in a Transgenic Model of Alzheimer's Disease. J Neurosci 2005; 25(17): 4217-21.
42- Lu B, Chow A. Neurotrophins and Hippocampal Synaptic Transmission and Plasticity. J Neurosci Res 1999; 58(1): 76-87.
43- Całyniuk B, Grochowska-Niedworok E, Walkiewicz KW, Kawecka S, Popiołek E, Fatyga E, et al. Malondialdehyde (MDA)–Product of Lipid Peroxidation as Marker of Homeostasis Disorders and Aging. Annales Acad Med Silesiensis 2016; 70: 224-8
44- Gil L, Siems W, Mazurek B, Gross J, Schroeder P, Voss P, et al. Age-Associated Analysis of Oxidative Stress Parameters in Human Plasma and Erythrocytes. Free Radic Res 2006; 40(5): 495-505.
45- Suresh D, Kumaran S, Annam V, Veena H. Age Related Changes in Malondialdehyde: Total Antioxidant Capacity Ratio-A Novel Marker of Oxidative Stress. Int J Pharm Bio Sci 2010; 1(2): 1-6.
46- Dei R, Takeda A, Niwa H, Li M, Nakagomi Y, Watanabe M, et al. Lipid Peroxidation and Advanced Glycation End Products in the Brain in Normal Aging and in Alzheimer's Disease. Acta Neuropathol 2002; 104(2): 113-22.
47- Dabidi-Roshan V, Hosseinzadeh S, Mahjoub S, Hosseinzadeh M, Myers J. Endurance Exercise Training and Diferuloyl Methane Supplement: Changes in Neurotrophic Factor and Oxidative Stress Induced by Lead in Rat Brain. Biol Sport 2013; 30(1): 41-6.
48- Fallah-Mohamadi Z, Hajizade-Moghadam A, Mahjob S, Azizi GH, Hoseini RS. Interaction Effect of Continuous Endurance Training and Homosistein Injection on Lipid Peroxidation and Antioxidant System in Male Rat Brain. Exerc Physiol 2011; 9(3): 117-28. [Persian]
49- Jacobsen Kt, Iverfeldt K. Amyloid Precursor Protein and its Homologues: A Family of Proteolysis-Dependent Receptors. Cell Mol Life Sci 2009; 66(14): 2299-318.
50- Prediger RD, Franco JL, Pandolfo P, Medeiros R, Duarte FS, Di Giunta G, et al. Differential Susceptibility Following β-Amyloid Peptide-(1–40) Administration in C57bl/6 and Swiss Albino Mice: Evidence for a Dissociation Between Cognitive Deficits and the Glutathione System Response. Behav Brain Res 2007; 177(2): 205-13.
51- Wang J, Ho L, Zhao W, Ono K, Rosensweig C, Chen L, et al. Grape-Derived Polyphenolics Prevent Aβ Oligomerization and Attenuate Cognitive Deterioration in a Mouse Model of Alzheimer's Disease. J Neurosci 2008; 28(25): 6388-92.
52- Shimmyo Y, Kihara T, Akaike A, Niidome T, Sugimoto H. Epigallocatechin-3-Gallate and Curcumin Suppress Amyloid Beta-Induced Beta-Site APP Cleaving Enzyme-1 Upregulation. Neuroreport 2008; 19(13): 1329-33.
53- Rybak L, Somani S, Ravi R. Effect of Exercise Training on Antioxidant System in Brain Regions of Rat. Pharmacol Biochem Behav 1995; 50(4): 635-9.
54- Cho JY, Um HS, Kang EB, Cho IH, Kim CH, Cho JS, Hwang DY. The Combination of Exercise Training and α-Lipoic Acid Treatment has Therapeutic Effects on the Pathogenic Phenotypes of Alzheimer's Disease in NSE/APPsw-Transgenic Mice. Int J Mol Med 2010; 25(3): 337-46.
55- Um HS, Kang EB, Leem YH, Cho IH, Yang CH, Chae KR, et al. Exercise Training Acts as a Therapeutic Strategy for Reduction of the Pathogenic Phenotypes for Alzheimer's Disease in an NSE/APPsw-Transgenic Model. Int J Mol Med 2008; 22(4): 529-39.
56- Baker LD, Frank LL, Foster-Schubert K, Green PS, Wilkinson CW, Mctiernan A, et al. Aerobic Exercise Improves Cognition for Older Adults with Glucose Intolerance, A Risk Factor for Alzheimer’s Disease. J Alzheimers Dis: JAD 2010; 22(2): 569-76.
1- Levin O, Fujiyama H, Boisgontier MP, Swinnen SP, Summers J. Aging and Motor Inhibition: A Converging Perspective Provided by Brain Stimulation and Imaging Approaches. Neurosci Biobehav Rev 2014; 43: 100-17.
2- Alexander GE, Ryan L, Bowers D, Foster TC, Bizon JL, Geldmacher DS, et al. Characterizing Cognitive Aging in Humans with Links to Animal Models. Front Aging Neurosci 2012; 4: 21.
3- Mendonca GV, Pezarat-Correia P, Vaz JR, Silva L, Heffernan KS. Impact of Aging on Endurance and Neuromuscular Physical Performance: The Role of Vascular Senescence. Sports Med 2017; 47(4): 583-98.
4- Khaledi S, Ahmadi S. Amyloid Beta and Tau: From Physiology to Pathology in Alzheimer’s Disease. Neurosci J Shefaye Khatam 2016; 4(4): 67-88 .[Persian]
5- Selkoe DJ. Alzheimer's Disease: Genotypes, Phenotype, and Treatments. Science 1997; 275(5300): 630.
6- Gispen WH, Biessels GJ. Cognition and Synaptic Plasticity in Diabetes Mellitus. Trends Neurosci 2000; 23(11): 542-9.
7- Zafra F, Lindholm D, Castren E, Hartikka J, Thoenen H. Regulation of Brain-Derived Neurotrophic Factor and Nerve Growth Factor Mrna in Primary Cultures of Hippocampal Neurons and Astrocytes. J Neurosci 1992; 12(12): 4793-9.
8- Dries DR, Yu G. Assembly, Maturation, And Trafficking of the γ-Secretase Complex in Alzheimer’s Disease. Curr Alzheimer Res 2008; 5(2): 132-46.
9- Sojkova J, Zhou Y, An Y, Kraut MA, Ferrucci L, Wong DF, et al. Longitudinal Patterns of β-Amyloid Deposition in Nondemented Older Adults. Arch Neurol 2011; 68(5): 644-9.
10- Butterfield DA, Sultana R. Methionine-35 of Aβ (1–42): Importance for Oxidative Stress in Alzheimer Disease. J Amino Acids 2011; 2011: 198430.
11- Glabe CC. Amyloid Accumulation and Pathogensis of Alzheimer’s Disease: Significance of Monomeric, Oligomeric and Fibrillar Aβ. Subcell Biochem 2005; 38: 167-77.
12- Caspersen C, Wang N, Yao J, Sosunov A, Chen X, Lustbader JW, et al. Mitochondrial Abeta: A Potential Focal Point for Neuronal Metabolic Dysfunction in Alzheimer's Disease. FASEB J 2005; 19(14): 2040-1.
13- Boyd-Kimball D, Mohmmad Abdul H, Reed T, Sultana R, Butterfield DA. Role of Phenylalanine 20 Inalzheimer's Amyloid β-Peptide (1-42)-Induced Oxidative Stress and Neurotoxicity. Chem Res Toxicol 2004; 17(12): 1743-9.
14- Nishida Y, Ito S, Ohtsuki S, Yamamoto N, Takahashi T, Iwata N, et al. Depletion of Vitamin E Increases Amyloid β Accumulation by Decreasing its Clearances from Brain and Blood in a Mouse Model of Alzheimer Disease. J Biol Chem 2009; 284(48): 33400-8.
15- Casado A, López-Fernández ME, Casado MC, de La Torre R. Lipid Peroxidation and Antioxidant Enzymeactivities in Vascular and Alzheimer Dementias. Neurochem Res 2008; 33(3): 450-8.
16- Berchtold N, Chinn G, Chou M, Kesslak J, Cotman C. Exercise Primes a Molecular Memory for Brain-Derived Neurotrophic Factor Protein Induction in the Rat Hippocampus. Neurosci 2005; 133(3): 853-61.
17- Friedland RP, Fritsch T, Smyth KA, Koss E, Lerner AJ, Chen CH, et al. Patients with Alzheimer's Disease Have Reduced Activities in Midlife Compared with Healthy Control-Group Members. Proc Natl Acad Sci Usa 2001; 98(6): 3440-5.
18- Laurin D, Verreault R, Lindsay J, Macpherson K, Rockwood K. Physical Activity and Risk of Cognitive Impairment and Dementia in Elderly Persons. Arch Neurol 2001; 58(3): 498-504.
19- Yu F, Xu B, Song C, Ji L, Zhang X. Treadmill Exercise Slows Cognitive Deficits in Aging Rats by Antioxidation and Inhibition of Amyloid Production. Neurorep 2013; 24(6): 342-7.
20- Zhao G, Liu H, Zhang H, Tong X. Treadmill Exercise Enhances Synaptic Plasticity, But Does Not Alter β-Amyloid Deposition in Hippocampi of Aged APP/PS1 Transgenic Mice. Neurosci 2015; 298: 357-66.
21- Yaghoubi A, Saghebjoo M, Fallah Mohammadi Z, Hedayati M, Hajizadeh-Moghaddam A. Effects of Continuous Training Intensity on Amyloid Beta1-42 (Aβ1-42) Levels in Hippocampus of Homocysteine-Induced Alzheimer's Model Rats. J Arak Uni Med Sci 2016; 18(11): 83-93.[Persian]
22- Seifert T, Brassard P, Wissenberg M, Rasmussen P, Nordby P, Stallknecht B, et al. Endurance Training Enhances BDNF Release from the Human Brain. Am J Physiol Regul Integr Comp Physiol 2010; 298(2): R372-7.
23- Groover AL, Ryals JM, Guilford BL, Wilson NM, Christianson JA, Wright DE. Exercise-Mediated Improvements in Painful Neuropathy Associated with Prediabetes in Mice. Pain 2013; 154(12): 2658-67.
24- Ziaaldini MM, Koltai E, Csende Z, Goto S, Boldogh I, Taylor AW, et al. Exercise Training Increases Anabolic and Attenuates Catabolic and Apoptotic Processes in Aged Skeletal Muscle of Male Rats. Exp Gerontol 2015; 67: 9-14.
25- Hafstad AD, Lund J, Hadler-Olsen E, Höper AC, Larsen TS, Aasum E. High- and Moderate-Intensity Training Normalizes Ventricular Function and Mechanoenergetics in Mice with Diet-Inducedobesity. Diabetes 2013; 62(7): 2287-94.
26- Hafstad AD, Boardman NT, Lund J, Hagve M, Khalid AM, Wisløff U, et al. High Intensity Interval Training Alters Substrate Utilization and Reduces Oxygen Consumption in The Heart. J Appl Physiol 2011; 111(5): 1234-41.
27- Azali Alamdari K, Khalafi M. The Effects of High Intensity Interval Training on Serum Levels of FGF21 and Insulin Resistance in Obese Men. IJDM 2019; 18(1): 41-8.[Persian]
28- Viola KL, Klein WL. Amyloid Βoligomers in Alzheimer’s Disease Pathogenesis, Treatment, and Diagnosis. Acta Neuropathol 2015; 129(2): 183-206.
29- Selkoe DJ, Hardy J. The Amyloid Hypothesis of Alzheimer's Disease at 25 Years. EMBO Mol Med 2016; 8(6): 595-608.
30- Uslu S, Akarkarasu ZE, Ozbabalik D, Ozkan S, Çolak O, Demirkan ES, et al. Levels of Amyloid Beta-42, Interleukin-6 and Tumor Necrosis Factor-Alpha in Alzheimer’s Disease and Vascular Dementia. Neurochem Res 2012; 37(7): 1554-9.
31- Capetillo-Zarate E, Gracia L, Tampellini D, Gouras GK. Intraneuronal Aβ Accumulation, Amyloid Plaques, And Synapse Pathology in Alzheimer’s Disease. Neurodegener Dis 2012; 10(1-4): 56-9.
32- Cavallucci V, D’amelio M, Cecconi F. Aβ Toxicity in Alzheimer's Disease. Mol Neurobiol 2012; 45(2): 366-78.
33- Souza LC, Carlos Filho B, Goes AT, Del Fabbro L, de Gomes MG, Savegnago L, et al. Neuroprotective Effect of Physical Exercise in a Mouse Model of Alzheimer’s Disease Induced by β-Amyloid1–40 Peptide. Neurotox Res 2013; 24(2): 148-63.
34- Paillard T, Rolland Y, de Souto Barreto P. Protective Effects of Physical Exercise in Alzheimer's Disease and Parkinson's Disease: A Narrative Review. J Clin Neurol 2015; 11(3): 212-9.
35- Kang EB, Cho JY. Effects of Treadmill Exercise on Brain Insulin Signaling and β-Amyloid in Intracerebroventricular Streptozotocin Induced-Memory Impairment in Rats. J Exerc Nutrition Biochem 2014; 18(1): 89-96.
36- Liu H-L, Zhao G, Zhang H, Shi LD. Long-Term Treadmill Exercise Inhibits the Progression of Alzheimer's Disease-Like Neuropathology in the Hippocampus of APP/PS1 Transgenic Mice. Behav Brain Res 2013; 256: 261-72.
37- Kang EB, Kwon IS, Koo JH, Kim EJ, Kim CH, Lee J, et al. Treadmill Exercise Represses Neuronal Cell Death and Inflammation During Aβ-Induced ER Stress by Regulating Unfolded Protein Response in Aged Presenilin 2 Mutant Mice. Apoptosis 2013; 18(11): 1332-47.
38- Cotman CW, Berchtold NC. Exercise: A Behavioral Intervention to Enhance Brain Health and Plasticity. Trends Nurosci 2002; 25(6): 295-301.
39- Radák Z, Sasvári M, Nyakas C, Taylor AW, Ohno H, Nakamoto H, et al. Regular Training Modulates the Accumulation of Reactive Carbonyl Derivatives in Mitochondrial and Cytosolic Fractions of Rat Skeletal Muscle. Arch Biochem Biophys 2000; 383(1): 114-8.
40- Schmitz A, Schneider A, Kummer MP, Herzog V. Endoplasmic Reticulum‐Localized Amyloid β‐Peptide is Degraded in the Cytosol by Two Distinct Degradation Pathways. Traffic 2004; 5(2): 89-101.
41- Adlard PA, Perreau VM, Pop V, Cotman CW. Voluntary Exercise Decreases Amyloid Load in a Transgenic Model of Alzheimer's Disease. J Neurosci 2005; 25(17): 4217-21.
42- Lu B, Chow A. Neurotrophins and Hippocampal Synaptic Transmission and Plasticity. J Neurosci Res 1999; 58(1): 76-87.
43- Całyniuk B, Grochowska-Niedworok E, Walkiewicz KW, Kawecka S, Popiołek E, Fatyga E, et al. Malondialdehyde (MDA)–Product of Lipid Peroxidation as Marker of Homeostasis Disorders and Aging. Annales Acad Med Silesiensis 2016; 70: 224-8
44- Gil L, Siems W, Mazurek B, Gross J, Schroeder P, Voss P, et al. Age-Associated Analysis of Oxidative Stress Parameters in Human Plasma and Erythrocytes. Free Radic Res 2006; 40(5): 495-505.
45- Suresh D, Kumaran S, Annam V, Veena H. Age Related Changes in Malondialdehyde: Total Antioxidant Capacity Ratio-A Novel Marker of Oxidative Stress. Int J Pharm Bio Sci 2010; 1(2): 1-6.
46- Dei R, Takeda A, Niwa H, Li M, Nakagomi Y, Watanabe M, et al. Lipid Peroxidation and Advanced Glycation End Products in the Brain in Normal Aging and in Alzheimer's Disease. Acta Neuropathol 2002; 104(2): 113-22.
47- Dabidi-Roshan V, Hosseinzadeh S, Mahjoub S, Hosseinzadeh M, Myers J. Endurance Exercise Training and Diferuloyl Methane Supplement: Changes in Neurotrophic Factor and Oxidative Stress Induced by Lead in Rat Brain. Biol Sport 2013; 30(1): 41-6.
48- Fallah-Mohamadi Z, Hajizade-Moghadam A, Mahjob S, Azizi GH, Hoseini RS. Interaction Effect of Continuous Endurance Training and Homosistein Injection on Lipid Peroxidation and Antioxidant System in Male Rat Brain. Exerc Physiol 2011; 9(3): 117-28. [Persian]
49- Jacobsen Kt, Iverfeldt K. Amyloid Precursor Protein and its Homologues: A Family of Proteolysis-Dependent Receptors. Cell Mol Life Sci 2009; 66(14): 2299-318.
50- Prediger RD, Franco JL, Pandolfo P, Medeiros R, Duarte FS, Di Giunta G, et al. Differential Susceptibility Following β-Amyloid Peptide-(1–40) Administration in C57bl/6 and Swiss Albino Mice: Evidence for a Dissociation Between Cognitive Deficits and the Glutathione System Response. Behav Brain Res 2007; 177(2): 205-13.
51- Wang J, Ho L, Zhao W, Ono K, Rosensweig C, Chen L, et al. Grape-Derived Polyphenolics Prevent Aβ Oligomerization and Attenuate Cognitive Deterioration in a Mouse Model of Alzheimer's Disease. J Neurosci 2008; 28(25): 6388-92.
52- Shimmyo Y, Kihara T, Akaike A, Niidome T, Sugimoto H. Epigallocatechin-3-Gallate and Curcumin Suppress Amyloid Beta-Induced Beta-Site APP Cleaving Enzyme-1 Upregulation. Neuroreport 2008; 19(13): 1329-33.
53- Rybak L, Somani S, Ravi R. Effect of Exercise Training on Antioxidant System in Brain Regions of Rat. Pharmacol Biochem Behav 1995; 50(4): 635-9.
54- Cho JY, Um HS, Kang EB, Cho IH, Kim CH, Cho JS, Hwang DY. The Combination of Exercise Training and α-Lipoic Acid Treatment has Therapeutic Effects on the Pathogenic Phenotypes of Alzheimer's Disease in NSE/APPsw-Transgenic Mice. Int J Mol Med 2010; 25(3): 337-46.
55- Um HS, Kang EB, Leem YH, Cho IH, Yang CH, Chae KR, et al. Exercise Training Acts as a Therapeutic Strategy for Reduction of the Pathogenic Phenotypes for Alzheimer's Disease in an NSE/APPsw-Transgenic Model. Int J Mol Med 2008; 22(4): 529-39.
56- Baker LD, Frank LL, Foster-Schubert K, Green PS, Wilkinson CW, Mctiernan A, et al. Aerobic Exercise Improves Cognition for Older Adults with Glucose Intolerance, A Risk Factor for Alzheimer’s Disease. J Alzheimers Dis: JAD 2010; 22(2): 569-76.
نوع مطالعه: پژوهشي |
موضوع مقاله:
فیزیولوژی ورزش
دریافت: 1399/12/7 | پذیرش: 1400/3/18 | انتشار: 1400/9/10
دریافت: 1399/12/7 | پذیرش: 1400/3/18 | انتشار: 1400/9/10
ارسال پیام به نویسنده مسئول
| بازنشر اطلاعات | |
 |
این مقاله تحت شرایط Creative Commons Attribution-NonCommercial 4.0 International License قابل بازنشر است. |
