

دوره ۲۹، شماره ۵ - ( مرداد ۱۴۰۰ )
جلد ۲۹ شماره ۵ صفحات ۳۷۰۹-۳۶۹۸ |
برگشت به فهرست نسخه ها


![]()
![]()
![]()
Download citation:
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:



BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
Parhamfar M, Rezaei M. Epigenetic Modifications of Host Genes Induced by Bacterial Infection. JSSU 2021; 29 (5) :3698-3709
URL: http://jssu.ssu.ac.ir/article-1-5179-fa.html
URL: http://jssu.ssu.ac.ir/article-1-5179-fa.html
پرهامفر مریم، رضایی مرضیه. اصلاحات اپیژنتیکی ناشی از عفونت باکتریایی در ژنهای میزبان. مجله علمي پژوهشي دانشگاه علوم پزشكي شهید صدوقی يزد. ۱۴۰۰; ۲۹ (۵) :۳۶۹۸-۳۷۰۹



متن کامل [PDF 991 kb]
(۷۵۳ دریافت)
| چکیده (HTML) (2032 مشاهده)
References:
1- Waddington CH. The epigenotype. Int J Epidemiol 2012; 41(1):10-3.
2- Van Speybroeckm L. From Epigenesis to Epigenetics: The Case of C.H. Waddington. Ann N Y Acad Sci 2002; 98: 61-81.
3- Casadesus J, Low D. Epigenetic Gene Regulation in the Bacterial World. Microbiol Mol Biol Rev 2006; 70 (3): 830-85.
4- Serio Tr, Cashikar Ag, Kowal As, Sawicki Gj, Lindquist Sl. Self-Perpetuating Changes in Sup35 Protein Conformation as a Mechanism of Heredity in Yeast. Biochem Soc Symp 2001; 68: 35-43.
5- Ferguson-Smith Ac, Surani Ma. Imprinting and the Epigenetic Asymmetry between Parental Genomes. Science 2001; 293(5532): 1086-89.
6- Wang Y, Fischle W, Cheung W, Jacobs S, Khorasanizadeh S, Allis CD. Beyond the Double Helix: Writing and Reading the Histone Code. Novartis Found Symp 2004; 259: 3-17.
7- Van Der Woude M, Hale Wb, Low Da. Formation of Dna Methylation Patterns: Nonmethylated Gatc Sequences in Gut and Pap Operons. J Bacteriol 1998; 180(22): 5913-20.
8- Levenson JM, Sweatt JD. Epigenetic Mechanisms in Memory Formation. Nat Rev Neurosci 2005; 6(2): 108-18.
9- True Hl, Berlin I, Lindquist Sl. Epigenetic Regulation of Translation Reveals Hidden Genetic Variation to Produce Complex Traits. Nature 2004; 431(7005): 184-7.
10- Satpute-Krishnan P, Serio Tr. Prion Protein Remodelling Confers an Immediate Phenotypic Switch. Nature 2005; 437 (7056): 262-5.
11- Van Der Woude Mw, Baumler Aj. Phase and Antigenic Variation in Bacteria. Clin Microbial Rev 2004; 17(3): 581-611.
12- Hernday A, Braaten B, Low D. The Intricate Workings of a Bacterial Epigenetic Switch. Adv Exp Med Biol 2004; 547: 83-9.
13- Feinberg Ap. Epigenetics at the Epicenter of Modern Medicine. JAMA 2008; 299(11): 1345-50.
14- Furrow RE, Christiansen B, Feldman MW. Environment sensitive Epigenetics and the Heritability of Complex Diseases. Genetics 2011; 189(4): 1377-87.
15- Hamon MA, Cossart P. Histone Modifications and Chromatin Remodeling During Bacterial Infections. Cell Host Microbe 2008; 4(2): 100-9.
16- Bhavsar A, Guttman J, Finlay B. Manipulation of Host-Cell Pathways by Bacterial Pathogens. Nature 2007; 449(7164): 827-61.
17- Dumas Me, Barton Rh, Toye A, Cloarec O, Blancher C, Rothwell A, et al. Metabolic Profiling Reveals a Contribution of Gut Micro biota to Fatty Liver Phenotype in Insulin-Resistant Mice. Proc Natl Acad Sci Usa 2006; 103(33): 12511-16.
18- Oka T, Sato H, Ouchida M, Utsunomiya A, Yoshino T. Cumulative Epigenetic Abnormalities in Host Genes with Viral and Microbial Infection during Initiation and Progression of Malignant Lymphoma/Leukemia. Cancer 2011; 3: 568-81.
19- Murata M, Azuma Y, Miura K, Rahman Ma, Matsutani M, Aoyama M, et al. Chlamydial Set Domain Protein Functions as a Histone Methyltransferase. Microbiology 2007; 153(Pt 2): 585-92.
20- Virgin HW, Wherry EJ, Ahmed R. Redefining chronic viral infection. Cell 2009; 138(1): 30-50.
21- Al Akeel R. Role of Epigenetic Reprogramming of Host Genes in Bacterial Pathogenesis. Saudi J Biol Sci 2013; 20(4): 305-9.
22- Hatami M, Fahimirad Sh, Parhamfar M. Epigenetic and its Applications. Arak: Arak University Publishers 2018: 20-26.
23- Grabiec Am, Potempa J. Epigenetic Regulation in Bacterial Infections: Targeting Histone Deacetylases. Crit Rev Microbiol 2018; 44(3): 336-50.
24- Baek Sh. When Signaling Kinases Meet Histones and Histone Modifiers in the Nucleus. Mol Cell 2011; 42(3): 274-84.
25- Opitz B, Puschel A, Beermann W, Hocke Ac, Forster S, Schmeck B, et al. Listeria Monocytogenes Activated P38 Mapk and Induced Il-8 Secretion in a Nucleotide-Binding Oligomerization Domain 1-Dependent Manner in Endothelial Cells. J Immunol 2006; 176: 484-90.
26- Schmeck B, Lorenz J, N’guessan Pd, Opitz B, Van Laak V, Zahlten J, et al. Histone Acetylation and Flagellin are Essential for Legionella Pneumophila-Induced Cytokine Expression. J Immunol 2008; 181(2): 940-47.
27- Saccani S, Pantano S, Natoli G. P38- Dependent Marking of Inflammatory Genes for Increased Nf-Κb Recruitment. Nat Immunol 2002; 3: 69-75.
28- Slevogt H, Schmeck B, Jonatat C, Zahlten J, Beermann W, Van Laak V, et al. Moraxella Catarrhalis Induces Inflammatory Response of Bronchial Epithelial Cells Via Mapk and Nf-Κb Activation and Histone Deacetylase Activity Reduction. Am J Physiol Lung Cell Mol Physiol 2006; 290(5): 818-26.
29- Haller D, Holt L, Kim Sc, Schwabe Rf, Sartor Rb, Jobin C. Transforming Growth Factor-Β1 Inhibits Non-Pathogenic Gram Negative Bacteria-Induced Nf-Κb Recruitment to the Interleukin-6 Gene Promoter in Intestinal Epithelial Cells Through Modulation of Histone Acetylation. J Biol Chem 2003; 278(26): 23851-860.
30- Bardwell AJ, Abdollahi M, Bardwell L. Anthrax Lethal Factor-Cleavage Products of Mapk (Mitogen-Activated Protein Kinase) Kinases Exhibit Reduced Binding to Their Cognate Mapks. Biochem J 2004; 378(Pt 2): 569-77.
31- Hamon Ma, Cossart P. K+ Efflux is required for Histone H3 Dephosphorylation by Listeria Monocytogenes Listeriolysin O and Other Pore-Forming Toxins. Infect Immun 2011; 79(7): 2839-46.
32- Pathak SK, Basu S, Bhattacharyya A, Pathak S, Banerjee A, Basu J, Kundu M. Tlr4-Dependent Nf-Κb Activation and Mitogen- And Stress-Activated Protein Kinase 1-Triggered Phosphorylation Events are Central to Helicobacter Pylori Peptidyl Prolyl Cis-, Trans-Isomerase (Hp0175)-Mediated Induction of Il-6 Release from Macrophages. J Immunol 2006; 177(11): 7950-58.
33- Xia G, Schneider-Stock R, Diestel A, Habold C, Krueger S, Roessner A, et al. Helicobacter Pylori Regulates P21 (Waf1) by Histone H4 Acetylation. Biochem Biophys Res Commun 2008; 369(2): 526-31.
34- Imai K, Ochiai K, Okamoto T. Reactivation of Latent Hiv-1 Infection by the Periodontopathic Bacterium Porphyromonas Gingivalis Involves Histone Modification. J Immunol 2009; 182(6): 3688-95.
35- Chen C, Wang L, Chen S, Wu X, Gu M, Chen X, et al. Convergence of Dna Methylation and Phosphorothioation Epigenetics in Bacterial Genomes. Proc Natl Acad Sci USA 2017; 114(17): 4501-6.
36- Maier J, Mohrle R, Jeltsch A. Design of Synthetic Epigenetic Circuits Featuring Memory Effects and Reversible Switching Based on Dna Methylation. Nat Commun 2017; 8: 15336.
37- Khaleghi M, Parhamfar M. Lactobacillus as Probiotic. Kerman: Shahid Bahonar University of Kerman; 2017: 43-50.
38- Ushijima T, Hattori N. Molecular Pathways: Involvement of Helicobacter Pylori-Triggered Inflammation in the Formation of an Epigenetic Field Defect, And Its Usefulness as Cancer Risk and Exposure Markers. Clin Cancer Res 2012; 18(4): 923-29.
39- Chan Aoo, Lam Sk, Wong Bc, Wong Wm, Yuen Mf, Yeung Yh, et al. Promoter Methylation of E-Cadherin Gene in Gastric Mucosa Associated with Helicobacter Pylori Infection and in Gastric Cancer. Gut 2003; 52(4): 502-6.
40- Yan J, Zhang M, Zhang J, Chen X, Zhang X. Helicobacter Pylori Infection Promotes Methylation of Wwox Gene In Human Gastric Cancer. Biochem Biophys Res Commun 2011; 408(1): 99-102.
41- Yao Y, Tao H, Park Di, Sepulveda Jl, Sepulveda Ar. Demonstration and Characterization of Mutations Induced by Helicobacter Pylori Organisms in Gastric Epithelial Cells. Helicobacter 2006; 11(4): 272-86.
42- Ando T, Yoshida T, Enomoto S, Asada K, Tatematsu M, Ichinose M, et al. Dna Methylation of Microrna Genes in Gastric Mucosae of Gastric Cancer Patients: Its Possible Involvement in the Formation of Epigenetic Field Defect. Int J Cancer 2009; 124(10): 2367-74.
43- Niwa T, Tsukamoto T, Toyoda T, Mori A, Tanaka H, Maekita T, et al. Inflammatory Processes Triggered by Helicobacter Pylori Infection Cause Aberrant Dna Methylation in Gastric Epithelial Cells. Cancer Res 2010; 70(4): 1430-40.
44- Hattori N, Ushijima T. Epigenetic Impact of Infection on Carcinogenesis: Mechanisms and Applications. Genome Med 2016; 8:10.
45- Tolg C, Sabha N, Cortese R, Panchal T, Ahsan A, Soliman A, et al. Coli Infection Provokes Epigenetic Downregulation of Cdkn2a (P16ink4a) in Uroepithelial Cells. Lab Invest 2011; 91(6): 825-36.
46- Berger Sl, Kouzarides T, Shiekhattar R, Shilatifard A. An Operational Definition of Epigenetics. Genes Dev 2009; 23(7): 781-83.
47- Chiariotti L, Coretti L, Pero R, Lembo F. Epigenetic Alterations Induced by Bacterial Lipopolysaccharides. Adv Exp Med Biol 2016; 879: 91-105.
48- Chen X, Yoza Bk, El Gazzar M, Hu Jy, Cousart Sl, Mccall Ce. Relb Sustains Iκbα Expression During Endotoxin Tolerance. Clin Vaccine Immunol 2009; 16: 104-10.
49- Nakajima T, Enomoto S, Yamashita S, Ando T, Nakanishi Y, Nakazawa K, et al. Persistence of a Component of Dna Methylation in Gastric Mucosae after Helicobacter Pylori Eradication. J Gastroenterol 2010; 45: 37-44.
50- Katoh M. Dysregulation of Stem Cell Signaling Network Due to Germline Mutation, Snp, Helicobacter Pylori Infection, Epigenetic Change and Genetic Alteration in Gastric Cancer. Cancer Biol Ther 2007; 6(6): 832-9.
51- Sun J. Enteric Bacteria and Cancer Stem Cells. Cancers (Basel) 2010; 3: 285-97.
متن کامل: (۳۰۴۲ مشاهده)
مقدمه
اپیژنتیک در باکتریها
در گذشته، اصطلاح "اپیژنتیک" برای توصیف تمایز سلولهای ژنتیکی یکسان به سمت انواع سلولهای مجزا به شکل بافت و اندام در طول توسعه یک ارگانیسم چند سلولی بهکار می6رفت. در سال 1942، وادینگتون برای اولین بار مفهوم اپیژنتیک را بهصورت تاثیر عوامل زیست محیطی در توسعه و ایجاد صفات خاص از طریق تعامل ژن - محیط، معرفی نمود (1). عبارت وادینگتون "تعامل ژن با محیط خود که باعث ایجاد فنوتیپ میشود" کلیدی برای زیست شناسی تکاملی میباشد، یعنی "ایدهای که خصوصیات فنوتیپی یا مورفولوژیکی و عملکردی یک ارگانیسم، بهطور مداوم با یک برنامه تعریف شده توسط ژنوم تحت تاثیر محیط ارگانیسم، بهوجود میآید" (2). در مفهوم گستردهتر، هر نوع اطلاعات اضافی بر روی توالی DNA (بهعنوان مثال متیلاسیون DNA) را میتوان "اپیژنتیک" در نظر گرفت (3). جنبههای مدرن اپیژنتیک به تغییر DNA یا پروتئینهای مرتبط بدون تغییر در توالی DNA، اشاره میکند که اطلاعات موجود را به نسل بعدی منتقل مینماید (شکل 1).
.jpg)
شکل 1: یک منطقه نماینده از DNA ژنومی نشان داده شده است. متیلاسیون باقی مانده سیتوزین با خاموش کردن ژن در ارتباط است. همچنین تغییرات در متیلاسیون میتواند اکتسابی باشد (برای مثال در سلولهای سرطانی). رونویسی ژن سرکوبگر تومور مرتبط با یک جزیره CpG هیپو متیله است (با رنگ قرمز نشان داده شده است) (3).
در مجموع پدیده اپیژنتیک شامل موارد زیر است:
• پریون¬ها که در ساختار پروتئین بهطور وراثتی منتقل میشوند (4)،
• نقش¬پذیری ژنی (genomic imprinting) که با سرکوب مونو آلل ژن مادری و یا پدری به ارث میرسد (5)،
• تغییر هیستون، مانند متیلاسیون لیزینها توسط فاز هیستون متیل ترانسفرازها (MTases) که حالت فعال و خاموش کروماتین را حفظ میکند (6)،
• تشکیل الگو¬های متیلاسیون DNA بهعنوان یک نتیجه از مهار متیلاسیون DNA اختصاصی بهوسیله اتصال پروتئین (7). هر کدام از این پدیدهها شامل هم حالتهای خود پایدار بوده که پروتئین یا DNA به آنها مرتبط هستند (8) و هم وضعیت خاصی از مولکولها میباشد که بر بیان ژن تاثیر میگذارند. تنظیم اپیژنتیک میتواند موجودات تک سلولی را قادر به پاسخ سریع به تنشهای محیطی و یا پیامدهی کند. بهعنوان مثال، مخمر پریون + PSIکه با تغییر ساختار فاکتور انتهایی ترجمه Sup35p تولید میشود، توسط سلولهای دختر به ارث میرسد. در پریون +PSI، کدونهای غیر کدگذار خوانده میشوند که می¬تواند یک مزیت بقا در شرایط نامطلوب مانند رشد در پاراکوات یا کافئین فراهم کند (9). در یک جمعیت مخمر، برخی از سلولهای مخمر، پریون را حمل می-کنند و برخی دیگر حمل نخواهند کرد. این وضعیت، انعطاف پذیری بالقوه در پاسخ جمعیت مخمر به تغییرات محیطی، فراهم میکند (10). باکتریها همچنین از مکانیسمهای اپیژنتیک برای تغییر فاز و کنترل این تغییر استفاده میکنند. در تغییر فاز، بیان ژن بین حالت فعال (فاز ON) و غیر فعال (فاز OFF) تناوب دارد. بهعنوان مثال، سلولهای اشرشیاکلی عامل عفونت دستگاه ادراری (UPEC) دستخوش تغییر فاز پیلی میباشند (شکل 2) (11). همچنین اغلب ژنهای ادهسین در اشرشیاکلی توسط مکانیسمهای اپیژنتیک درگیر در الگوهای متیلاسیون DNA تنظیم میشوند (12).
1. تغییرات میزبان ناشی از پاتوژن
غالباً تصور بر این است که بیماریها عمدتاً توسط تغییرات ژنتیکی اکتسابی، هدایت میشوند. در حال حاضر، روشن شده است که هر فنوتیپ نتیجه فعل و انفعالات پیچیده بین ژنوتیپ، اپی-ژنوم و محیط زیست میباشد (13). متیلاسیون DNA و هیستون، استیلاسیون، فسفریلاسیون، ADP- ریبوزیلاسیون، تکرار ناشی از ژنهای خاموش، تداخل miRNAها، تجمع، نقشپذیری ژنی و غیره ...، تعداد کمی از نمونههای اپیژنتیک میباشند (14،13). از طرفی تغییرات فیزیولوژی، مورفولوژی و رفتاری میزبان ناشی از پاتوژن، به طور گسترده در تحقیقات علمی نشان داده شده است. یکی از جذابترین نمونههای این تغییرات، آنهایی هستند که نتیجه یک استراتژی دستکاری پاتوژن هدف در به حداکثر رساندن بقا و انتقال را نشان دادهاند. شواهدی گزارش شده که تغییرات هیستون و بازسازی کروماتین، بیان ژن را تنظیم میکند که اهداف کلیدی برای دستکاری پاتوژن در طول یک عفونت هستند (15). از اهداف دیگر، سیستم ایمنی بدن میزبان است. مدولاسیون اپیژنتیک در برنامه رونویسی میزبان به ژنهای دفاعی میزبان مرتبط میشود که معمولاً در عفونت باکتریایی و حتی ویروسی، آشکار می گردد (16).
2. تغییرات اپیژنتیک ناشی از تهاجم میکروبی
مطالعات متعددی نشان دادهاند که عوامل عفونی خاص (هلیکوباکتر پیلوری، استرپتوکوکوس بوویس، کلامیدیا پنومونیه، کمپیلوباکتر رکتوس، ویروس اپشتینبار، ویروس هپاتیت، ویروس پاپیلومای انسانی، پولیوما ویروس و غیره) میتوانند در تغییرات اپیژنتیک از جمله متیلاسیون ژن میزبان، مشارکت کنند و منجر به شروع و پیشرفت برخی از بیماریها بهخصوص سرطان شوند. بهعنوان مثال، نقش فلور میکروبی روده پستانداران بهعنوان فاکتور تغییر دهنده اپیژنتیک در بیماری سندرم متابولیک و سایر بیماریهای مرتبط گزارش شده است (17). به این ترتیب، سیگنالهای مختلف زنده و غیر زنده باعث تغییر در بیان ژن میشوند و حتی میتوانند پس از توقف اثر، باقی بمانند (18). علاوه براین، باکتریهای بومی روده در مکانیسمهای اپیژنومی و عواقب ناشی از عدم تعادل میکرواکولوژیک روده و ناهنجاریهای اپیژنتیک در شروع و پیشرفت بیماری نقش دارند (19). میکروبهایی که باعث عفونتهای مداوم میشوند، احتمالاً از تغییرات اپیژنتیک ارثی در رونویسی میزبان بهره میبرند که برای حالت نهفته یا مزمن خود، بدون نیاز به بیان مداوم عوامل موثر در شروع و راه اندازی یک محیط ویژه، فعالیت میکنند (20). برخی از عفونتهای باکتریایی مزمن نیز با بدخیمی همراه هستند که در این زمینه، بیشتر عفونت هلیکوباکتر پیلوری در مخاط معده انسان مورد مطالعه قرار گرفته است (21). علاوه بر باکتریها، عفونتهای ویروسی نیز میتوانند باعث عدم تنظیم الگوهای سرکوبگر تغییرات هیستونی شوند که می¬تواند متیلاسیون DNA نابجا، برنامهریزی مجدد سلولهای آلوده و نسل بعدی آنها را تسریع کند. این مسئله میتواند باعث مهار ژنهای سرکوبگر تومور شود که یک انتخاب قوی در پیشرفت سرطان خواهد بود (20). در نهایت باید متذکر شد که مکانیسمهای کنترل بیان ژن در سطح کروماتین، سطح بالایی از پیچیدگی را نشان میدهند. محصولات باکتریایی مختلف میتوانند آنها را در بسیاری از مسیرها (جدول 1) از طریق فعالسازی سیگنالهای متوالی یا بهطور مستقیم در هسته، تحت تاثیر قرار دهند که در ادامه به تفصیل آمده است (22).
جدول 1: گونههای باکتریایی، عوامل و اثرات
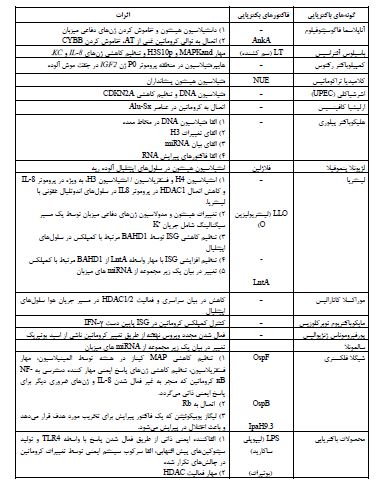
3. گونههای باکتریایی، عوامل و اثرات
3-1. تغییرات هیستونی
تاکنون، بسیاری از تغییرات کروماتین القا شده توسط باکتریها مانند، استیلاسیون / داستیلاسیون و فسفریلاسیون / دفسفریلاسیون هیستون از طریق فعال شدن پیامدهی آبشاری سلول میزبان توسط اجزای باکتریایی (بهعنوان مثال، الگوهای مولکولی وابسته به میکروب، متابولیتها و عوامل ویرولانس) رخ میدهند (23). در میان مسیرهای سیگنالدهی میزبان که تعدادی از باکتریها فعال میشوند، MAPKs، NF-kB و مسیرهای PI3K برای فعال کردن کینازها که H3S10 را در هسته فسفریله میکنند، شناخته شده¬اند (24). هر محرک باکتریایی فعال در این مسیرها، دارای پتانسیل القا H3S10p میباشد و مرتبط با هیستونهای استیله شده است. در سلول-های HUVEC اندوتلیال، لیستریا مونوسیتوژنز عامل لیستریوز به سرعت فسفریلاسیون و استیلاسیون هیستون¬ها، به ویژه در پروموتر ژن پیش التهابیIL-8 را افزایش میدهد (25). فلاژل لژیونلا پنموفیلا، عامل بیماری لژیونر و لیپوپلیساکارید باکتریایی (LPS)، دارای اثرات مشابهی به ترتیب روی IL-8 در سلولهای اپیتلیال ریه و سلولهای دندریتیک میباشند (27، 26). باکتریهای همسفره، مانند موراکسلا کاتارالیس، یک باکتری ساپروفیت دستگاه تنفسی یا باکتریوئیدس ولگاتوس یک کامنسال فلور رودهای، میتوانند فسفریلاسیون/ استیلاسیون H3 را از طریق القا یک پیامدهی آبشاری التهابی، تحریک کنند (29 ،28). سموم باکتریایی مانند سم کشنده (LT) باسیلوس آنتراسیس عامل سیاهزخم، نیز پاسخ ایمنی ذاتی میزبان را توسط مهار فسفریلاسیون/ استیلاسیون H3، کاهش میدهند (30). سموم دیگر، از جمله لیستریولیزین O (LLO) لیستریا مونوسیتوژنز، PFO کلستریدیوم پرفرینجنس،PLY استرپتوکوکوس پنومونیه و آئرولیزین از آئروموناس هیدروفیلا اثرات مشابهی دارند (31). باکتری هلیکوباکتر پیلوری سرطانزا نیز قادر به تغییر هیستون با استفاده از ابزارهای متنوع از جمله مدولاسیون مسیر MAPK است (32). تغییرات کروماتین ممکن است به اثرات هلیکوباکتر در پیشرفت چرخه سلولی، تکثیر و مرگ سلولها کمک کند (33). باکتریها همچنین میتوانند متابولیتهای فعال بهعنوان مهار کنندههای آنزیمهای تغییر دهنده کروماتین تولید کنند که یکی از این محصولات اسید بوتیریک میباشد. قابل ذکر است که اثر سوء پورفیروموناس ژنژیوالیس در فعال شدن مجدد ویروسهای پنهان، مانند ویروس نقص ایمنی انسانی (HIV) و سندرم اپشتین بار (EBV)، ظاهراً ناشی از تولید بوتیرات توسط این باکتری میباشد (34).
3-2. متیلاسیون DNA
اکثر سیستمهای اپیژنتیک شناخته شده در باکتریها از متیلاسیون DNA بهعنوان یک سیگنال استفاده میکنند که تعامل DNA - پروتئین خاص را تنظیم می¬کند. این سیستمها معمولا از یک DNA آدنین متیلاز (Dam) و پروتئینهای متصل شونده به DNA تشکیل شده¬اند که این پروتئینها به توالیهایی از DNA که با جایگاه متیلاسیون هدف همپوشانی دارد، متصل شده و مکان متیلاسیون آن محل را مسدود میکند. متیلاسیون جایگاه هدف به نوبه خود، مانع از اتصال پروتئین می¬شود و منجر به دو حالت متیلاسیون متناوب (متیله و غیرمتیله) در جایگاه هدف میشود (35). دو مورد از آنزیمهای متیلاز،Dam متیلاز از باکتریهای رودهای و CcrM متیلاز از کالوباکتر کرسنتوس میباشند که نمونههای یک فرآیند تکاملی بوده که در متیلاسیون آدنین DNA بهعنوان یک مکانیسم سیگنالدهی فعالیت می¬کنند و تعاملات DNA - پروتئین را تنظیم می¬نمایند. در هر دو گاما و آلفاپروتئوباکتریا، متیلاسیون آدنین DNA، همانندسازی کروموزوم و رونویسی جفت ژنهای خاص برای عبور چنگال همانندسازی DNA را تنظیم میکند. در برخی موارد، پروتئین متصل شونده تنظیم کننده، متیلاسیون DNA را مهار میکند و الگوهای متیلاسیون DNA که نشانه وضعیت¬های متناوب اپی¬ژنتیک هستند، تولید می¬کند. الگو¬های متیلاسیون DNA توسط شرایط محیطی از طریق تغییرات در پروتئین متصل شونده نظارتی، تعدیل می-شوند. جمعیت¬های باکتریایی ممکن است از الگوهای متیلاسیون DNA به ارث برده، استفاده کنند که بهعنوان یک حافظه کوتاه مدت شرایط متابولیک میباشد که در نسل قبل، پیشرفت کرده و تقسیم می¬شوند. متیلاسیون DNA نیز نقش مهمی در تنوع پاتوژن¬های باکتریایی دارد و احتمال طراحی دارو¬های ضد باکتری جدید را بالا می¬برد که ممکن است متیلاسیون آدنین DNA را مهار کند. از یک دارو از این نوع می¬توان انتظار داشت که ویرولانس نوع وحشی باکتری¬ها را توسط تبدیل آنها به فنوکپیهای موتانهای Dam، مهار کند (36). باید در نظر داشت که با افزایش چشمگیر باکتریهای مقاوم به آنتیبیوتیک، امروزه طراحی و معرفی داروهای جدید، اهمیت قابل توجهی دارد (37). نقش وقوع متیلاسیون DNA در ارتباط با عفونتهای باکتریایی بهطور فزاینده درک شده است. بهترین مثال، عفونت هلیکوباکتر پیلوری میباشد که باعث متیلاسیون DNA نابجا در مخاط معده انسان میشود و بهطور قابل توجهی در پروموتر¬های ژن¬های متیله در سلولهای سرطان معده، یافت شده است (38). هایپرمتیلاسیون وابسته بههلیکوباکتر پیلوری، برای مثال در ژن E–کادهرین در CDH1 (39)، ژن سرکوب کننده تومور (40)، ژن ترمیم کننده DNA (41) و همچنین در جزایر CpG در ژن¬های miRNA ها رخ می¬دهد (42). توانایی هلیکوباکتر پیلوری به القاء متیلاسیون DNA در مخاط معده در مدل حیوانی موش صحرائی تایید شده است (43). همچنین، در برابر محرک اتانول یاNaCl که موجب نفوذ نوتروفیل¬ها در معده می¬شود، التهاب با واسطه هلیکوباکتر پیلوری باعث رها شدن لنفوسیت-ها و نفوذ ماکروفاژ¬ها می¬گردد که به نظر می¬رسد نقش کلیدی در القای متیلاسیون داشته باشد (شکل 3) (44). علاوه براین، متیلاسیون DNA القا شده توسط باکتری می¬تواند بر ژن¬های دخیل در تکثیر سلولی و بقا پاتوژن، تاثیر گذارد، مانند آنچه توسط اشرشیاکلای عامل عفونت ادراری (UPEC) نشان داده شده است (45). همچنین برخی از مطالعات نشان داده¬اند که کمپیلوباکتر رکتوس درگیر در عفونتهای پریودنتال و مرتبط با افزایش خطر تولد زودرس ناشی از عفونت جفت و جنین در انسان میتواند در موش آلوده، متیلاسیون بیش از حد در منطقه پروموتر P0 از ژن IGF2 در جفت (فاکتور رشد 2 شبه انسولین) را القا کند (46). بنابراین محدودیت رشد داخل رحمی که در حیوانات آلوده به کمپیلوباکتر رکتوس مشاهده شده، یک پیامد از تغییر اپی¬ژنتیک ناشی از عفونت باکتریایی در جفت موش می باشد (21).
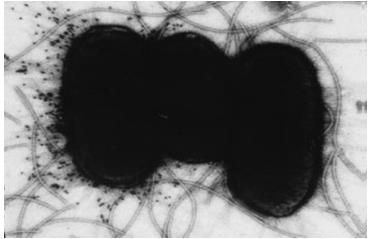
شکل 2: تغییر فاز پیلی Pap17 در اشرشیاکلی سویه C1212 در مبتلایان به عفونت ادراری که با آنتیبادیهای آنتی- Pap17 نشاندار شده با ذرات 10 نانومتری کلوئیدی طلا قابل مشاهده است. باکتری در سمت چپ در حالت فاز ON برای بیان Pap17 است، در حالیکه دو باکتری در سمت راست در فاز OFF هستند. لازم به ذکر است که این دو باکتری، پیلی Pap21 غیرنشاندار را بیان میکنند که تحت کنترل تغییر فاز هستند اما با آنتیسرم آنتی- Pap17 مشخص نمیشوند (11).

شکل 3: مکانیسم¬های القاء متیلاسیون DNA نابجا توسط عفونت هلیکوباکتر پیلوری (44).
4. حافظه اپیژنتیک عفونت: نقشپذیری ژنی باکتریایی
تغییرات کروماتین ممکن است به سلولهای دختر در طول تقسیم سلولی منتقل شود و منجر به تغییرات وراثتی در عملکرد ژن گردد، همچنین احتمال دارد یک عفونت باکتریایی علائم قابل توارث پس از ریشهکن کردن پاتوژن تولید کند. دو مثال زیر از این ایده حمایت می کنند.
4-1. اجزای اپی¬ژنتیک التهاب سیستمیک شدید و تحمل LPS
واکنش¬های التهابی نابجا در پاسخ به قرار گرفتن مداوم در معرض میکروبها و محصولات میکروبی مانند LPS، منجر به آسیب بافت، اختلال عملکرد چند اندام، شوک سپتیک و مرگ می¬شوند. برای جبران این عوارض جانبی، سیستم ایمنی، مکانیسم سرکوب ایمنی پس از عفونت (postseptic immunosuppression :PSI) را توسعه داده که سلول¬های خونساز قادر به تبدیل شدن به سلولهای واکنشی ضعیف موقتی می¬شوند. این پاسخ ضد التهابی جبرانی با اثرات مضر عفونت، مقابله مینماید، اما افراد برای مدت زمان طولانی (از هفته تا سال) به عفونتهای فرصت طلب حساس باقی می-مانند (47). یکی از جنبههای PSI، تحمل LPS است که در آن پاسخهای TLR4 حاصل از LPS به سمت خاموشی ژن¬های سیتوکین پیش التهابی و بیان واسطه¬های ضد میکروبی یا ضد التهابی، برنامهریزی میشوند. باز شدن کروماتین در ژنهای هدف در این فاز حاد، شامل فسفریلاسیون هیستون و استیلاسیون می¬باشد (48).
4-2. سرطانهای مرتبط با عفونت باکتریایی
همانطور که قبلا ذکر شد، هلیکوباکتر پیلوری یک عامل خطر اکتسابی مهم برای سرطان معده است. این باکتری دارای پروتئین و آنتیژنهای مختلفی (CagA, FlaA, VacA) میباشد که در بیماریزایی آن نقش عمدهای دارند و بررسیهای متعددی برای شناسایی و تولید نوترکیب و تعیین آنتی¬ژنیسیتی آنها صورت گرفته است (40-38). علاوه بر این، اثرات با واسطه هلیکوباکتر پیلوری بر رشد سلول و تمامیت DNA ناشی از متیلاسیون DNA نابجا بهعنوان یک مکانیسم مهم در بروز سرطان معده، ظاهر شده است (44). شایان ذکر است که ریشه کن کردن عفونت هلیکوباکتر پیلوری در بیماران انسانی باعث ناپدید شدن کامل متیلاسیون پروموتر جزایر CpG در ژنهای نزدیک مرتبط با خطر ایجاد سرطان معده نمی¬شود، اما این فرایند را کاهش میدهد (49). این یک نشانه قوی است که یک عفونت باکتریایی ممکن است آثار اپیژنتیک در یک بافت باقی بگذارد که منجر به تغییرات دائمی در بیان ژن شود. با توجه به این واقعیت که سرطان باید از یک سلول ایجاد شود که دارای پتانسیل تقسیم میباشد، این برنامهریزی مجدد باکتری ممکن است در سلول¬های با عمر طولانی، مانند سلولهای بنیادی یا سلولهای اجدادی، القا شود و در نتیجه به سلول¬های دختر گسترش یابد. همچنین ممکن است سلول¬های اپیتلیال بالغ برای عدم تمایز با اجزای خاموش، شبکه پیامدهی سلولهای بنیادی را مورد هدف قرار دهد که منجر به افزایش تکثیر و بقای آنها میشود (50). "نمونه هلیکوباکتر"، قابلیت جابجایی به دیگر بافت¬های هدف باکتری را نیز دارد. همچنین این باور وجود دارد که ممکن است عفونت باکتری اشرشیاکلی با خطر ابتلا به سرطان مثانه در ارتباط باشد (45) و باکتریهای روده¬ای مستعد کننده سرطان روده بزرگ باشند. بهطور کلی، عدم تنظیم سرکوب کننده تومور و یا مسیرهای مرتبط با سلولهای بنیادی (بهعنوان مثال Wnt، JAK-STAT، JNK و Notch) در تغییر ژنتیکی و برنامهریزی مجدد اپیژنتیک ناشی از باکتری، یک دلیل احتمالی از توسعه سرطان در نیچ¬های اپیتلیالی است (51).
نتیجهگیری
تنظیم ژنوم پیچیده یوکاریوتها نه تنها مستلزم فاکتورهای متصل شونده بهتوالی DNA اختصاصی است، بلکه به سطوح بیشتری از تنظیمات از قبیل تغییرات DNA، تغییرات هیستون پس از ترجمه و بازسازی کروماتین، نیاز دارد. این تغییرات اغلب بهعنوان اپی¬ژنتیک نشان داده می¬شوند. توانایی میکروب-ها که بهطور اپی¬ژنتیکی بیان ژن میزبان را تحت تاثیر قرار میدهند، نقش عمده¬ای در پاتوژنز بیماریهای مزمن دارند. شواهد رو به رشدی برای نقش عفونت باکتریایی در مدولاسیون اطلاعات اپیژنتیک سلولهای میزبان توسط مکانیسم¬های متنوع وجود دارد. امروزه پیشرفت¬های فنآوری، نقشه برداری متیلاسیون DNA ژنوم کامل انسان و پروفایل تغییرات هیستونی را امکانپذیر کرده است. باکتری¬هایی که باعث تغییرات کروماتین در سلول¬های پستانداران می¬شوند، بدون شک از این تکنولوژی¬های جدید بهرهمند میشوند. اپی-ژنتیک در تنظیم رونویسی، تقسیم سلولی، ترمیم و تکثیر DNA نقش مهمی دارد. از دست دادن کنترل اپیژنتیک از این فرآیندهای مبتنی بر DNA ممکن است در ارتباط با بیماری-های عفونی باکتریایی باشد. برای درک نقش تغییرات کروماتین و تنظیم کننده¬های دیگر در بیماری¬های عفونی، تحقیقات باید در سطح بافت و سلول¬های دارای عمر طولانی انجام شود. نتایج این تحقیقات احتمالاً به نوع سلول و زمینه ژنتیکی میزبان بستگی دارد. با درک بهتر ارتباط بین بیماری-های عفونی باکتریایی و اپی¬ژنوم، فرصت برای کاربرد¬های درمانی به خصوص ایجاد ترکیبات دارویی موثرتر و برگرداندن فرآیندهای اپیژنتیک، بهوجود می¬آید. همچنین حذف سریع تغییرات پاتو- اپی¬ژنتیک ناشی از میکروب¬ها ممکن است از عفونتهای مزمن و یا پنهان، برخی سرطانها یا بیماریهای خود ایمنی، پیشگیری کند.
سپاسگزاری
بدین وسیله نویسندگان، مراتب تشکر و قدردانی خود را از خانم دکتر شهره فهیمیراد به علت طرح ایده و همکاری در بحث اعلام میدارند.
حامی مالی: ندارد.
تعارض در منافع: وجود ندارد.
اپیژنتیک در باکتریها
در گذشته، اصطلاح "اپیژنتیک" برای توصیف تمایز سلولهای ژنتیکی یکسان به سمت انواع سلولهای مجزا به شکل بافت و اندام در طول توسعه یک ارگانیسم چند سلولی بهکار می6رفت. در سال 1942، وادینگتون برای اولین بار مفهوم اپیژنتیک را بهصورت تاثیر عوامل زیست محیطی در توسعه و ایجاد صفات خاص از طریق تعامل ژن - محیط، معرفی نمود (1). عبارت وادینگتون "تعامل ژن با محیط خود که باعث ایجاد فنوتیپ میشود" کلیدی برای زیست شناسی تکاملی میباشد، یعنی "ایدهای که خصوصیات فنوتیپی یا مورفولوژیکی و عملکردی یک ارگانیسم، بهطور مداوم با یک برنامه تعریف شده توسط ژنوم تحت تاثیر محیط ارگانیسم، بهوجود میآید" (2). در مفهوم گستردهتر، هر نوع اطلاعات اضافی بر روی توالی DNA (بهعنوان مثال متیلاسیون DNA) را میتوان "اپیژنتیک" در نظر گرفت (3). جنبههای مدرن اپیژنتیک به تغییر DNA یا پروتئینهای مرتبط بدون تغییر در توالی DNA، اشاره میکند که اطلاعات موجود را به نسل بعدی منتقل مینماید (شکل 1).
شکل 1: یک منطقه نماینده از DNA ژنومی نشان داده شده است. متیلاسیون باقی مانده سیتوزین با خاموش کردن ژن در ارتباط است. همچنین تغییرات در متیلاسیون میتواند اکتسابی باشد (برای مثال در سلولهای سرطانی). رونویسی ژن سرکوبگر تومور مرتبط با یک جزیره CpG هیپو متیله است (با رنگ قرمز نشان داده شده است) (3).
در مجموع پدیده اپیژنتیک شامل موارد زیر است:
• پریون¬ها که در ساختار پروتئین بهطور وراثتی منتقل میشوند (4)،
• نقش¬پذیری ژنی (genomic imprinting) که با سرکوب مونو آلل ژن مادری و یا پدری به ارث میرسد (5)،
• تغییر هیستون، مانند متیلاسیون لیزینها توسط فاز هیستون متیل ترانسفرازها (MTases) که حالت فعال و خاموش کروماتین را حفظ میکند (6)،
• تشکیل الگو¬های متیلاسیون DNA بهعنوان یک نتیجه از مهار متیلاسیون DNA اختصاصی بهوسیله اتصال پروتئین (7). هر کدام از این پدیدهها شامل هم حالتهای خود پایدار بوده که پروتئین یا DNA به آنها مرتبط هستند (8) و هم وضعیت خاصی از مولکولها میباشد که بر بیان ژن تاثیر میگذارند. تنظیم اپیژنتیک میتواند موجودات تک سلولی را قادر به پاسخ سریع به تنشهای محیطی و یا پیامدهی کند. بهعنوان مثال، مخمر پریون + PSIکه با تغییر ساختار فاکتور انتهایی ترجمه Sup35p تولید میشود، توسط سلولهای دختر به ارث میرسد. در پریون +PSI، کدونهای غیر کدگذار خوانده میشوند که می¬تواند یک مزیت بقا در شرایط نامطلوب مانند رشد در پاراکوات یا کافئین فراهم کند (9). در یک جمعیت مخمر، برخی از سلولهای مخمر، پریون را حمل می-کنند و برخی دیگر حمل نخواهند کرد. این وضعیت، انعطاف پذیری بالقوه در پاسخ جمعیت مخمر به تغییرات محیطی، فراهم میکند (10). باکتریها همچنین از مکانیسمهای اپیژنتیک برای تغییر فاز و کنترل این تغییر استفاده میکنند. در تغییر فاز، بیان ژن بین حالت فعال (فاز ON) و غیر فعال (فاز OFF) تناوب دارد. بهعنوان مثال، سلولهای اشرشیاکلی عامل عفونت دستگاه ادراری (UPEC) دستخوش تغییر فاز پیلی میباشند (شکل 2) (11). همچنین اغلب ژنهای ادهسین در اشرشیاکلی توسط مکانیسمهای اپیژنتیک درگیر در الگوهای متیلاسیون DNA تنظیم میشوند (12).
1. تغییرات میزبان ناشی از پاتوژن
غالباً تصور بر این است که بیماریها عمدتاً توسط تغییرات ژنتیکی اکتسابی، هدایت میشوند. در حال حاضر، روشن شده است که هر فنوتیپ نتیجه فعل و انفعالات پیچیده بین ژنوتیپ، اپی-ژنوم و محیط زیست میباشد (13). متیلاسیون DNA و هیستون، استیلاسیون، فسفریلاسیون، ADP- ریبوزیلاسیون، تکرار ناشی از ژنهای خاموش، تداخل miRNAها، تجمع، نقشپذیری ژنی و غیره ...، تعداد کمی از نمونههای اپیژنتیک میباشند (14،13). از طرفی تغییرات فیزیولوژی، مورفولوژی و رفتاری میزبان ناشی از پاتوژن، به طور گسترده در تحقیقات علمی نشان داده شده است. یکی از جذابترین نمونههای این تغییرات، آنهایی هستند که نتیجه یک استراتژی دستکاری پاتوژن هدف در به حداکثر رساندن بقا و انتقال را نشان دادهاند. شواهدی گزارش شده که تغییرات هیستون و بازسازی کروماتین، بیان ژن را تنظیم میکند که اهداف کلیدی برای دستکاری پاتوژن در طول یک عفونت هستند (15). از اهداف دیگر، سیستم ایمنی بدن میزبان است. مدولاسیون اپیژنتیک در برنامه رونویسی میزبان به ژنهای دفاعی میزبان مرتبط میشود که معمولاً در عفونت باکتریایی و حتی ویروسی، آشکار می گردد (16).
2. تغییرات اپیژنتیک ناشی از تهاجم میکروبی
مطالعات متعددی نشان دادهاند که عوامل عفونی خاص (هلیکوباکتر پیلوری، استرپتوکوکوس بوویس، کلامیدیا پنومونیه، کمپیلوباکتر رکتوس، ویروس اپشتینبار، ویروس هپاتیت، ویروس پاپیلومای انسانی، پولیوما ویروس و غیره) میتوانند در تغییرات اپیژنتیک از جمله متیلاسیون ژن میزبان، مشارکت کنند و منجر به شروع و پیشرفت برخی از بیماریها بهخصوص سرطان شوند. بهعنوان مثال، نقش فلور میکروبی روده پستانداران بهعنوان فاکتور تغییر دهنده اپیژنتیک در بیماری سندرم متابولیک و سایر بیماریهای مرتبط گزارش شده است (17). به این ترتیب، سیگنالهای مختلف زنده و غیر زنده باعث تغییر در بیان ژن میشوند و حتی میتوانند پس از توقف اثر، باقی بمانند (18). علاوه براین، باکتریهای بومی روده در مکانیسمهای اپیژنومی و عواقب ناشی از عدم تعادل میکرواکولوژیک روده و ناهنجاریهای اپیژنتیک در شروع و پیشرفت بیماری نقش دارند (19). میکروبهایی که باعث عفونتهای مداوم میشوند، احتمالاً از تغییرات اپیژنتیک ارثی در رونویسی میزبان بهره میبرند که برای حالت نهفته یا مزمن خود، بدون نیاز به بیان مداوم عوامل موثر در شروع و راه اندازی یک محیط ویژه، فعالیت میکنند (20). برخی از عفونتهای باکتریایی مزمن نیز با بدخیمی همراه هستند که در این زمینه، بیشتر عفونت هلیکوباکتر پیلوری در مخاط معده انسان مورد مطالعه قرار گرفته است (21). علاوه بر باکتریها، عفونتهای ویروسی نیز میتوانند باعث عدم تنظیم الگوهای سرکوبگر تغییرات هیستونی شوند که می¬تواند متیلاسیون DNA نابجا، برنامهریزی مجدد سلولهای آلوده و نسل بعدی آنها را تسریع کند. این مسئله میتواند باعث مهار ژنهای سرکوبگر تومور شود که یک انتخاب قوی در پیشرفت سرطان خواهد بود (20). در نهایت باید متذکر شد که مکانیسمهای کنترل بیان ژن در سطح کروماتین، سطح بالایی از پیچیدگی را نشان میدهند. محصولات باکتریایی مختلف میتوانند آنها را در بسیاری از مسیرها (جدول 1) از طریق فعالسازی سیگنالهای متوالی یا بهطور مستقیم در هسته، تحت تاثیر قرار دهند که در ادامه به تفصیل آمده است (22).
جدول 1: گونههای باکتریایی، عوامل و اثرات
3. گونههای باکتریایی، عوامل و اثرات
3-1. تغییرات هیستونی
تاکنون، بسیاری از تغییرات کروماتین القا شده توسط باکتریها مانند، استیلاسیون / داستیلاسیون و فسفریلاسیون / دفسفریلاسیون هیستون از طریق فعال شدن پیامدهی آبشاری سلول میزبان توسط اجزای باکتریایی (بهعنوان مثال، الگوهای مولکولی وابسته به میکروب، متابولیتها و عوامل ویرولانس) رخ میدهند (23). در میان مسیرهای سیگنالدهی میزبان که تعدادی از باکتریها فعال میشوند، MAPKs، NF-kB و مسیرهای PI3K برای فعال کردن کینازها که H3S10 را در هسته فسفریله میکنند، شناخته شده¬اند (24). هر محرک باکتریایی فعال در این مسیرها، دارای پتانسیل القا H3S10p میباشد و مرتبط با هیستونهای استیله شده است. در سلول-های HUVEC اندوتلیال، لیستریا مونوسیتوژنز عامل لیستریوز به سرعت فسفریلاسیون و استیلاسیون هیستون¬ها، به ویژه در پروموتر ژن پیش التهابیIL-8 را افزایش میدهد (25). فلاژل لژیونلا پنموفیلا، عامل بیماری لژیونر و لیپوپلیساکارید باکتریایی (LPS)، دارای اثرات مشابهی به ترتیب روی IL-8 در سلولهای اپیتلیال ریه و سلولهای دندریتیک میباشند (27، 26). باکتریهای همسفره، مانند موراکسلا کاتارالیس، یک باکتری ساپروفیت دستگاه تنفسی یا باکتریوئیدس ولگاتوس یک کامنسال فلور رودهای، میتوانند فسفریلاسیون/ استیلاسیون H3 را از طریق القا یک پیامدهی آبشاری التهابی، تحریک کنند (29 ،28). سموم باکتریایی مانند سم کشنده (LT) باسیلوس آنتراسیس عامل سیاهزخم، نیز پاسخ ایمنی ذاتی میزبان را توسط مهار فسفریلاسیون/ استیلاسیون H3، کاهش میدهند (30). سموم دیگر، از جمله لیستریولیزین O (LLO) لیستریا مونوسیتوژنز، PFO کلستریدیوم پرفرینجنس،PLY استرپتوکوکوس پنومونیه و آئرولیزین از آئروموناس هیدروفیلا اثرات مشابهی دارند (31). باکتری هلیکوباکتر پیلوری سرطانزا نیز قادر به تغییر هیستون با استفاده از ابزارهای متنوع از جمله مدولاسیون مسیر MAPK است (32). تغییرات کروماتین ممکن است به اثرات هلیکوباکتر در پیشرفت چرخه سلولی، تکثیر و مرگ سلولها کمک کند (33). باکتریها همچنین میتوانند متابولیتهای فعال بهعنوان مهار کنندههای آنزیمهای تغییر دهنده کروماتین تولید کنند که یکی از این محصولات اسید بوتیریک میباشد. قابل ذکر است که اثر سوء پورفیروموناس ژنژیوالیس در فعال شدن مجدد ویروسهای پنهان، مانند ویروس نقص ایمنی انسانی (HIV) و سندرم اپشتین بار (EBV)، ظاهراً ناشی از تولید بوتیرات توسط این باکتری میباشد (34).
3-2. متیلاسیون DNA
اکثر سیستمهای اپیژنتیک شناخته شده در باکتریها از متیلاسیون DNA بهعنوان یک سیگنال استفاده میکنند که تعامل DNA - پروتئین خاص را تنظیم می¬کند. این سیستمها معمولا از یک DNA آدنین متیلاز (Dam) و پروتئینهای متصل شونده به DNA تشکیل شده¬اند که این پروتئینها به توالیهایی از DNA که با جایگاه متیلاسیون هدف همپوشانی دارد، متصل شده و مکان متیلاسیون آن محل را مسدود میکند. متیلاسیون جایگاه هدف به نوبه خود، مانع از اتصال پروتئین می¬شود و منجر به دو حالت متیلاسیون متناوب (متیله و غیرمتیله) در جایگاه هدف میشود (35). دو مورد از آنزیمهای متیلاز،Dam متیلاز از باکتریهای رودهای و CcrM متیلاز از کالوباکتر کرسنتوس میباشند که نمونههای یک فرآیند تکاملی بوده که در متیلاسیون آدنین DNA بهعنوان یک مکانیسم سیگنالدهی فعالیت می¬کنند و تعاملات DNA - پروتئین را تنظیم می¬نمایند. در هر دو گاما و آلفاپروتئوباکتریا، متیلاسیون آدنین DNA، همانندسازی کروموزوم و رونویسی جفت ژنهای خاص برای عبور چنگال همانندسازی DNA را تنظیم میکند. در برخی موارد، پروتئین متصل شونده تنظیم کننده، متیلاسیون DNA را مهار میکند و الگوهای متیلاسیون DNA که نشانه وضعیت¬های متناوب اپی¬ژنتیک هستند، تولید می¬کند. الگو¬های متیلاسیون DNA توسط شرایط محیطی از طریق تغییرات در پروتئین متصل شونده نظارتی، تعدیل می-شوند. جمعیت¬های باکتریایی ممکن است از الگوهای متیلاسیون DNA به ارث برده، استفاده کنند که بهعنوان یک حافظه کوتاه مدت شرایط متابولیک میباشد که در نسل قبل، پیشرفت کرده و تقسیم می¬شوند. متیلاسیون DNA نیز نقش مهمی در تنوع پاتوژن¬های باکتریایی دارد و احتمال طراحی دارو¬های ضد باکتری جدید را بالا می¬برد که ممکن است متیلاسیون آدنین DNA را مهار کند. از یک دارو از این نوع می¬توان انتظار داشت که ویرولانس نوع وحشی باکتری¬ها را توسط تبدیل آنها به فنوکپیهای موتانهای Dam، مهار کند (36). باید در نظر داشت که با افزایش چشمگیر باکتریهای مقاوم به آنتیبیوتیک، امروزه طراحی و معرفی داروهای جدید، اهمیت قابل توجهی دارد (37). نقش وقوع متیلاسیون DNA در ارتباط با عفونتهای باکتریایی بهطور فزاینده درک شده است. بهترین مثال، عفونت هلیکوباکتر پیلوری میباشد که باعث متیلاسیون DNA نابجا در مخاط معده انسان میشود و بهطور قابل توجهی در پروموتر¬های ژن¬های متیله در سلولهای سرطان معده، یافت شده است (38). هایپرمتیلاسیون وابسته بههلیکوباکتر پیلوری، برای مثال در ژن E–کادهرین در CDH1 (39)، ژن سرکوب کننده تومور (40)، ژن ترمیم کننده DNA (41) و همچنین در جزایر CpG در ژن¬های miRNA ها رخ می¬دهد (42). توانایی هلیکوباکتر پیلوری به القاء متیلاسیون DNA در مخاط معده در مدل حیوانی موش صحرائی تایید شده است (43). همچنین، در برابر محرک اتانول یاNaCl که موجب نفوذ نوتروفیل¬ها در معده می¬شود، التهاب با واسطه هلیکوباکتر پیلوری باعث رها شدن لنفوسیت-ها و نفوذ ماکروفاژ¬ها می¬گردد که به نظر می¬رسد نقش کلیدی در القای متیلاسیون داشته باشد (شکل 3) (44). علاوه براین، متیلاسیون DNA القا شده توسط باکتری می¬تواند بر ژن¬های دخیل در تکثیر سلولی و بقا پاتوژن، تاثیر گذارد، مانند آنچه توسط اشرشیاکلای عامل عفونت ادراری (UPEC) نشان داده شده است (45). همچنین برخی از مطالعات نشان داده¬اند که کمپیلوباکتر رکتوس درگیر در عفونتهای پریودنتال و مرتبط با افزایش خطر تولد زودرس ناشی از عفونت جفت و جنین در انسان میتواند در موش آلوده، متیلاسیون بیش از حد در منطقه پروموتر P0 از ژن IGF2 در جفت (فاکتور رشد 2 شبه انسولین) را القا کند (46). بنابراین محدودیت رشد داخل رحمی که در حیوانات آلوده به کمپیلوباکتر رکتوس مشاهده شده، یک پیامد از تغییر اپی¬ژنتیک ناشی از عفونت باکتریایی در جفت موش می باشد (21).
شکل 2: تغییر فاز پیلی Pap17 در اشرشیاکلی سویه C1212 در مبتلایان به عفونت ادراری که با آنتیبادیهای آنتی- Pap17 نشاندار شده با ذرات 10 نانومتری کلوئیدی طلا قابل مشاهده است. باکتری در سمت چپ در حالت فاز ON برای بیان Pap17 است، در حالیکه دو باکتری در سمت راست در فاز OFF هستند. لازم به ذکر است که این دو باکتری، پیلی Pap21 غیرنشاندار را بیان میکنند که تحت کنترل تغییر فاز هستند اما با آنتیسرم آنتی- Pap17 مشخص نمیشوند (11).
شکل 3: مکانیسم¬های القاء متیلاسیون DNA نابجا توسط عفونت هلیکوباکتر پیلوری (44).
4. حافظه اپیژنتیک عفونت: نقشپذیری ژنی باکتریایی
تغییرات کروماتین ممکن است به سلولهای دختر در طول تقسیم سلولی منتقل شود و منجر به تغییرات وراثتی در عملکرد ژن گردد، همچنین احتمال دارد یک عفونت باکتریایی علائم قابل توارث پس از ریشهکن کردن پاتوژن تولید کند. دو مثال زیر از این ایده حمایت می کنند.
4-1. اجزای اپی¬ژنتیک التهاب سیستمیک شدید و تحمل LPS
واکنش¬های التهابی نابجا در پاسخ به قرار گرفتن مداوم در معرض میکروبها و محصولات میکروبی مانند LPS، منجر به آسیب بافت، اختلال عملکرد چند اندام، شوک سپتیک و مرگ می¬شوند. برای جبران این عوارض جانبی، سیستم ایمنی، مکانیسم سرکوب ایمنی پس از عفونت (postseptic immunosuppression :PSI) را توسعه داده که سلول¬های خونساز قادر به تبدیل شدن به سلولهای واکنشی ضعیف موقتی می¬شوند. این پاسخ ضد التهابی جبرانی با اثرات مضر عفونت، مقابله مینماید، اما افراد برای مدت زمان طولانی (از هفته تا سال) به عفونتهای فرصت طلب حساس باقی می-مانند (47). یکی از جنبههای PSI، تحمل LPS است که در آن پاسخهای TLR4 حاصل از LPS به سمت خاموشی ژن¬های سیتوکین پیش التهابی و بیان واسطه¬های ضد میکروبی یا ضد التهابی، برنامهریزی میشوند. باز شدن کروماتین در ژنهای هدف در این فاز حاد، شامل فسفریلاسیون هیستون و استیلاسیون می¬باشد (48).
4-2. سرطانهای مرتبط با عفونت باکتریایی
همانطور که قبلا ذکر شد، هلیکوباکتر پیلوری یک عامل خطر اکتسابی مهم برای سرطان معده است. این باکتری دارای پروتئین و آنتیژنهای مختلفی (CagA, FlaA, VacA) میباشد که در بیماریزایی آن نقش عمدهای دارند و بررسیهای متعددی برای شناسایی و تولید نوترکیب و تعیین آنتی¬ژنیسیتی آنها صورت گرفته است (40-38). علاوه بر این، اثرات با واسطه هلیکوباکتر پیلوری بر رشد سلول و تمامیت DNA ناشی از متیلاسیون DNA نابجا بهعنوان یک مکانیسم مهم در بروز سرطان معده، ظاهر شده است (44). شایان ذکر است که ریشه کن کردن عفونت هلیکوباکتر پیلوری در بیماران انسانی باعث ناپدید شدن کامل متیلاسیون پروموتر جزایر CpG در ژنهای نزدیک مرتبط با خطر ایجاد سرطان معده نمی¬شود، اما این فرایند را کاهش میدهد (49). این یک نشانه قوی است که یک عفونت باکتریایی ممکن است آثار اپیژنتیک در یک بافت باقی بگذارد که منجر به تغییرات دائمی در بیان ژن شود. با توجه به این واقعیت که سرطان باید از یک سلول ایجاد شود که دارای پتانسیل تقسیم میباشد، این برنامهریزی مجدد باکتری ممکن است در سلول¬های با عمر طولانی، مانند سلولهای بنیادی یا سلولهای اجدادی، القا شود و در نتیجه به سلول¬های دختر گسترش یابد. همچنین ممکن است سلول¬های اپیتلیال بالغ برای عدم تمایز با اجزای خاموش، شبکه پیامدهی سلولهای بنیادی را مورد هدف قرار دهد که منجر به افزایش تکثیر و بقای آنها میشود (50). "نمونه هلیکوباکتر"، قابلیت جابجایی به دیگر بافت¬های هدف باکتری را نیز دارد. همچنین این باور وجود دارد که ممکن است عفونت باکتری اشرشیاکلی با خطر ابتلا به سرطان مثانه در ارتباط باشد (45) و باکتریهای روده¬ای مستعد کننده سرطان روده بزرگ باشند. بهطور کلی، عدم تنظیم سرکوب کننده تومور و یا مسیرهای مرتبط با سلولهای بنیادی (بهعنوان مثال Wnt، JAK-STAT، JNK و Notch) در تغییر ژنتیکی و برنامهریزی مجدد اپیژنتیک ناشی از باکتری، یک دلیل احتمالی از توسعه سرطان در نیچ¬های اپیتلیالی است (51).
نتیجهگیری
تنظیم ژنوم پیچیده یوکاریوتها نه تنها مستلزم فاکتورهای متصل شونده بهتوالی DNA اختصاصی است، بلکه به سطوح بیشتری از تنظیمات از قبیل تغییرات DNA، تغییرات هیستون پس از ترجمه و بازسازی کروماتین، نیاز دارد. این تغییرات اغلب بهعنوان اپی¬ژنتیک نشان داده می¬شوند. توانایی میکروب-ها که بهطور اپی¬ژنتیکی بیان ژن میزبان را تحت تاثیر قرار میدهند، نقش عمده¬ای در پاتوژنز بیماریهای مزمن دارند. شواهد رو به رشدی برای نقش عفونت باکتریایی در مدولاسیون اطلاعات اپیژنتیک سلولهای میزبان توسط مکانیسم¬های متنوع وجود دارد. امروزه پیشرفت¬های فنآوری، نقشه برداری متیلاسیون DNA ژنوم کامل انسان و پروفایل تغییرات هیستونی را امکانپذیر کرده است. باکتری¬هایی که باعث تغییرات کروماتین در سلول¬های پستانداران می¬شوند، بدون شک از این تکنولوژی¬های جدید بهرهمند میشوند. اپی-ژنتیک در تنظیم رونویسی، تقسیم سلولی، ترمیم و تکثیر DNA نقش مهمی دارد. از دست دادن کنترل اپیژنتیک از این فرآیندهای مبتنی بر DNA ممکن است در ارتباط با بیماری-های عفونی باکتریایی باشد. برای درک نقش تغییرات کروماتین و تنظیم کننده¬های دیگر در بیماری¬های عفونی، تحقیقات باید در سطح بافت و سلول¬های دارای عمر طولانی انجام شود. نتایج این تحقیقات احتمالاً به نوع سلول و زمینه ژنتیکی میزبان بستگی دارد. با درک بهتر ارتباط بین بیماری-های عفونی باکتریایی و اپی¬ژنوم، فرصت برای کاربرد¬های درمانی به خصوص ایجاد ترکیبات دارویی موثرتر و برگرداندن فرآیندهای اپیژنتیک، بهوجود می¬آید. همچنین حذف سریع تغییرات پاتو- اپی¬ژنتیک ناشی از میکروب¬ها ممکن است از عفونتهای مزمن و یا پنهان، برخی سرطانها یا بیماریهای خود ایمنی، پیشگیری کند.
سپاسگزاری
بدین وسیله نویسندگان، مراتب تشکر و قدردانی خود را از خانم دکتر شهره فهیمیراد به علت طرح ایده و همکاری در بحث اعلام میدارند.
حامی مالی: ندارد.
تعارض در منافع: وجود ندارد.
References:
1- Waddington CH. The epigenotype. Int J Epidemiol 2012; 41(1):10-3.
2- Van Speybroeckm L. From Epigenesis to Epigenetics: The Case of C.H. Waddington. Ann N Y Acad Sci 2002; 98: 61-81.
3- Casadesus J, Low D. Epigenetic Gene Regulation in the Bacterial World. Microbiol Mol Biol Rev 2006; 70 (3): 830-85.
4- Serio Tr, Cashikar Ag, Kowal As, Sawicki Gj, Lindquist Sl. Self-Perpetuating Changes in Sup35 Protein Conformation as a Mechanism of Heredity in Yeast. Biochem Soc Symp 2001; 68: 35-43.
5- Ferguson-Smith Ac, Surani Ma. Imprinting and the Epigenetic Asymmetry between Parental Genomes. Science 2001; 293(5532): 1086-89.
6- Wang Y, Fischle W, Cheung W, Jacobs S, Khorasanizadeh S, Allis CD. Beyond the Double Helix: Writing and Reading the Histone Code. Novartis Found Symp 2004; 259: 3-17.
7- Van Der Woude M, Hale Wb, Low Da. Formation of Dna Methylation Patterns: Nonmethylated Gatc Sequences in Gut and Pap Operons. J Bacteriol 1998; 180(22): 5913-20.
8- Levenson JM, Sweatt JD. Epigenetic Mechanisms in Memory Formation. Nat Rev Neurosci 2005; 6(2): 108-18.
9- True Hl, Berlin I, Lindquist Sl. Epigenetic Regulation of Translation Reveals Hidden Genetic Variation to Produce Complex Traits. Nature 2004; 431(7005): 184-7.
10- Satpute-Krishnan P, Serio Tr. Prion Protein Remodelling Confers an Immediate Phenotypic Switch. Nature 2005; 437 (7056): 262-5.
11- Van Der Woude Mw, Baumler Aj. Phase and Antigenic Variation in Bacteria. Clin Microbial Rev 2004; 17(3): 581-611.
12- Hernday A, Braaten B, Low D. The Intricate Workings of a Bacterial Epigenetic Switch. Adv Exp Med Biol 2004; 547: 83-9.
13- Feinberg Ap. Epigenetics at the Epicenter of Modern Medicine. JAMA 2008; 299(11): 1345-50.
14- Furrow RE, Christiansen B, Feldman MW. Environment sensitive Epigenetics and the Heritability of Complex Diseases. Genetics 2011; 189(4): 1377-87.
15- Hamon MA, Cossart P. Histone Modifications and Chromatin Remodeling During Bacterial Infections. Cell Host Microbe 2008; 4(2): 100-9.
16- Bhavsar A, Guttman J, Finlay B. Manipulation of Host-Cell Pathways by Bacterial Pathogens. Nature 2007; 449(7164): 827-61.
17- Dumas Me, Barton Rh, Toye A, Cloarec O, Blancher C, Rothwell A, et al. Metabolic Profiling Reveals a Contribution of Gut Micro biota to Fatty Liver Phenotype in Insulin-Resistant Mice. Proc Natl Acad Sci Usa 2006; 103(33): 12511-16.
18- Oka T, Sato H, Ouchida M, Utsunomiya A, Yoshino T. Cumulative Epigenetic Abnormalities in Host Genes with Viral and Microbial Infection during Initiation and Progression of Malignant Lymphoma/Leukemia. Cancer 2011; 3: 568-81.
19- Murata M, Azuma Y, Miura K, Rahman Ma, Matsutani M, Aoyama M, et al. Chlamydial Set Domain Protein Functions as a Histone Methyltransferase. Microbiology 2007; 153(Pt 2): 585-92.
20- Virgin HW, Wherry EJ, Ahmed R. Redefining chronic viral infection. Cell 2009; 138(1): 30-50.
21- Al Akeel R. Role of Epigenetic Reprogramming of Host Genes in Bacterial Pathogenesis. Saudi J Biol Sci 2013; 20(4): 305-9.
22- Hatami M, Fahimirad Sh, Parhamfar M. Epigenetic and its Applications. Arak: Arak University Publishers 2018: 20-26.
23- Grabiec Am, Potempa J. Epigenetic Regulation in Bacterial Infections: Targeting Histone Deacetylases. Crit Rev Microbiol 2018; 44(3): 336-50.
24- Baek Sh. When Signaling Kinases Meet Histones and Histone Modifiers in the Nucleus. Mol Cell 2011; 42(3): 274-84.
25- Opitz B, Puschel A, Beermann W, Hocke Ac, Forster S, Schmeck B, et al. Listeria Monocytogenes Activated P38 Mapk and Induced Il-8 Secretion in a Nucleotide-Binding Oligomerization Domain 1-Dependent Manner in Endothelial Cells. J Immunol 2006; 176: 484-90.
26- Schmeck B, Lorenz J, N’guessan Pd, Opitz B, Van Laak V, Zahlten J, et al. Histone Acetylation and Flagellin are Essential for Legionella Pneumophila-Induced Cytokine Expression. J Immunol 2008; 181(2): 940-47.
27- Saccani S, Pantano S, Natoli G. P38- Dependent Marking of Inflammatory Genes for Increased Nf-Κb Recruitment. Nat Immunol 2002; 3: 69-75.
28- Slevogt H, Schmeck B, Jonatat C, Zahlten J, Beermann W, Van Laak V, et al. Moraxella Catarrhalis Induces Inflammatory Response of Bronchial Epithelial Cells Via Mapk and Nf-Κb Activation and Histone Deacetylase Activity Reduction. Am J Physiol Lung Cell Mol Physiol 2006; 290(5): 818-26.
29- Haller D, Holt L, Kim Sc, Schwabe Rf, Sartor Rb, Jobin C. Transforming Growth Factor-Β1 Inhibits Non-Pathogenic Gram Negative Bacteria-Induced Nf-Κb Recruitment to the Interleukin-6 Gene Promoter in Intestinal Epithelial Cells Through Modulation of Histone Acetylation. J Biol Chem 2003; 278(26): 23851-860.
30- Bardwell AJ, Abdollahi M, Bardwell L. Anthrax Lethal Factor-Cleavage Products of Mapk (Mitogen-Activated Protein Kinase) Kinases Exhibit Reduced Binding to Their Cognate Mapks. Biochem J 2004; 378(Pt 2): 569-77.
31- Hamon Ma, Cossart P. K+ Efflux is required for Histone H3 Dephosphorylation by Listeria Monocytogenes Listeriolysin O and Other Pore-Forming Toxins. Infect Immun 2011; 79(7): 2839-46.
32- Pathak SK, Basu S, Bhattacharyya A, Pathak S, Banerjee A, Basu J, Kundu M. Tlr4-Dependent Nf-Κb Activation and Mitogen- And Stress-Activated Protein Kinase 1-Triggered Phosphorylation Events are Central to Helicobacter Pylori Peptidyl Prolyl Cis-, Trans-Isomerase (Hp0175)-Mediated Induction of Il-6 Release from Macrophages. J Immunol 2006; 177(11): 7950-58.
33- Xia G, Schneider-Stock R, Diestel A, Habold C, Krueger S, Roessner A, et al. Helicobacter Pylori Regulates P21 (Waf1) by Histone H4 Acetylation. Biochem Biophys Res Commun 2008; 369(2): 526-31.
34- Imai K, Ochiai K, Okamoto T. Reactivation of Latent Hiv-1 Infection by the Periodontopathic Bacterium Porphyromonas Gingivalis Involves Histone Modification. J Immunol 2009; 182(6): 3688-95.
35- Chen C, Wang L, Chen S, Wu X, Gu M, Chen X, et al. Convergence of Dna Methylation and Phosphorothioation Epigenetics in Bacterial Genomes. Proc Natl Acad Sci USA 2017; 114(17): 4501-6.
36- Maier J, Mohrle R, Jeltsch A. Design of Synthetic Epigenetic Circuits Featuring Memory Effects and Reversible Switching Based on Dna Methylation. Nat Commun 2017; 8: 15336.
37- Khaleghi M, Parhamfar M. Lactobacillus as Probiotic. Kerman: Shahid Bahonar University of Kerman; 2017: 43-50.
38- Ushijima T, Hattori N. Molecular Pathways: Involvement of Helicobacter Pylori-Triggered Inflammation in the Formation of an Epigenetic Field Defect, And Its Usefulness as Cancer Risk and Exposure Markers. Clin Cancer Res 2012; 18(4): 923-29.
39- Chan Aoo, Lam Sk, Wong Bc, Wong Wm, Yuen Mf, Yeung Yh, et al. Promoter Methylation of E-Cadherin Gene in Gastric Mucosa Associated with Helicobacter Pylori Infection and in Gastric Cancer. Gut 2003; 52(4): 502-6.
40- Yan J, Zhang M, Zhang J, Chen X, Zhang X. Helicobacter Pylori Infection Promotes Methylation of Wwox Gene In Human Gastric Cancer. Biochem Biophys Res Commun 2011; 408(1): 99-102.
41- Yao Y, Tao H, Park Di, Sepulveda Jl, Sepulveda Ar. Demonstration and Characterization of Mutations Induced by Helicobacter Pylori Organisms in Gastric Epithelial Cells. Helicobacter 2006; 11(4): 272-86.
42- Ando T, Yoshida T, Enomoto S, Asada K, Tatematsu M, Ichinose M, et al. Dna Methylation of Microrna Genes in Gastric Mucosae of Gastric Cancer Patients: Its Possible Involvement in the Formation of Epigenetic Field Defect. Int J Cancer 2009; 124(10): 2367-74.
43- Niwa T, Tsukamoto T, Toyoda T, Mori A, Tanaka H, Maekita T, et al. Inflammatory Processes Triggered by Helicobacter Pylori Infection Cause Aberrant Dna Methylation in Gastric Epithelial Cells. Cancer Res 2010; 70(4): 1430-40.
44- Hattori N, Ushijima T. Epigenetic Impact of Infection on Carcinogenesis: Mechanisms and Applications. Genome Med 2016; 8:10.
45- Tolg C, Sabha N, Cortese R, Panchal T, Ahsan A, Soliman A, et al. Coli Infection Provokes Epigenetic Downregulation of Cdkn2a (P16ink4a) in Uroepithelial Cells. Lab Invest 2011; 91(6): 825-36.
46- Berger Sl, Kouzarides T, Shiekhattar R, Shilatifard A. An Operational Definition of Epigenetics. Genes Dev 2009; 23(7): 781-83.
47- Chiariotti L, Coretti L, Pero R, Lembo F. Epigenetic Alterations Induced by Bacterial Lipopolysaccharides. Adv Exp Med Biol 2016; 879: 91-105.
48- Chen X, Yoza Bk, El Gazzar M, Hu Jy, Cousart Sl, Mccall Ce. Relb Sustains Iκbα Expression During Endotoxin Tolerance. Clin Vaccine Immunol 2009; 16: 104-10.
49- Nakajima T, Enomoto S, Yamashita S, Ando T, Nakanishi Y, Nakazawa K, et al. Persistence of a Component of Dna Methylation in Gastric Mucosae after Helicobacter Pylori Eradication. J Gastroenterol 2010; 45: 37-44.
50- Katoh M. Dysregulation of Stem Cell Signaling Network Due to Germline Mutation, Snp, Helicobacter Pylori Infection, Epigenetic Change and Genetic Alteration in Gastric Cancer. Cancer Biol Ther 2007; 6(6): 832-9.
51- Sun J. Enteric Bacteria and Cancer Stem Cells. Cancers (Basel) 2010; 3: 285-97.
نوع مطالعه: مروری |
موضوع مقاله:
میکروبیولوژی
دریافت: 1399/4/14 | پذیرش: 1399/8/26 | انتشار: 1400/5/10
دریافت: 1399/4/14 | پذیرش: 1399/8/26 | انتشار: 1400/5/10
ارسال پیام به نویسنده مسئول
| بازنشر اطلاعات | |
 |
این مقاله تحت شرایط Creative Commons Attribution-NonCommercial 4.0 International License قابل بازنشر است. |
